{"title":"Bullous Vasculitis in Eosinophilic Granulomatosis with Polyangiitis: A Case Report.","authors":"Tiraporn Phumwiriya, Charussri Leeyaphan","doi":"10.1159/000544815","DOIUrl":null,"url":null,"abstract":"<p><strong>Introduction: </strong>Eosinophilic granulomatosis with polyangiitis (EGPA) is a rare systemic vasculitis affecting small- and medium-sized vessels. It is characterized by multiorgan involvement and can lead to severe outcomes if not diagnosed promptly. Cutaneous manifestations are common and typically include palpable purpura and subcutaneous nodules. Widespread bullous vasculitis affecting areas such as the forehead and ear presents an atypical presentation. We report a case of EGPA presenting with bullous vasculitis in an unusual location.</p><p><strong>Case presentation: </strong>A 40-year-old woman with a history of late-onset allergic rhinitis presented with a 2-week history of numbness in her right leg, along with multiple erythematous papules and vesicles, some with shallow erosions, located on the forehead and left ear. She also experienced fever, progressive dyspnea, and hemoptysis. She was diagnosed with pneumonitis, alveolar hemorrhage, and mononeuritis of the right leg. Laboratory findings revealed leukocytosis with eosinophilia, and the anti-myeloperoxidase antibody was positive. Histopathological examination of the bullous lesion on the forehead showed intraepidermal separation with necrotic keratinocytes and prominent eosinophil infiltration, along with focal leukocytoclastic vasculitis. The patient was diagnosed with EGPA and started on intravenous steroids and cyclophosphamide. EGPA is a rare disease characterized by multiorgan vasculitis, asthma, and granulomatous eosinophilic inflammation, which are its key hallmarks. While cutaneous involvement is common, bullous vasculitis is rarely observed on the forehead and ear.</p><p><strong>Conclusions: </strong>EGPA is a challenging diagnosis due to its variable presentation. While cutaneous manifestations are common, widespread bullous vasculitis may be atypical and rare clinical presentation. This case underscores the importance of considering EGPA in the differential diagnosis of bullous vasculitis, particularly when associated with systemic symptoms and eosinophilia. Early recognition and treatment are crucial for improving outcomes in this potentially life-threatening condition.</p>","PeriodicalId":9619,"journal":{"name":"Case Reports in Dermatology","volume":"17 1","pages":"74-79"},"PeriodicalIF":0.8000,"publicationDate":"2025-02-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11936433/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Case Reports in Dermatology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1159/000544815","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"DERMATOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
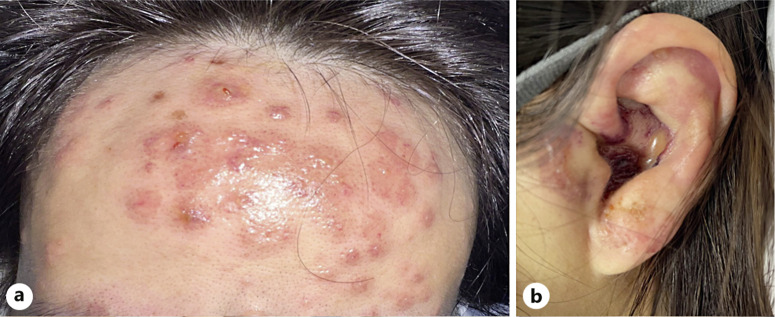
Introduction: Eosinophilic granulomatosis with polyangiitis (EGPA) is a rare systemic vasculitis affecting small- and medium-sized vessels. It is characterized by multiorgan involvement and can lead to severe outcomes if not diagnosed promptly. Cutaneous manifestations are common and typically include palpable purpura and subcutaneous nodules. Widespread bullous vasculitis affecting areas such as the forehead and ear presents an atypical presentation. We report a case of EGPA presenting with bullous vasculitis in an unusual location.
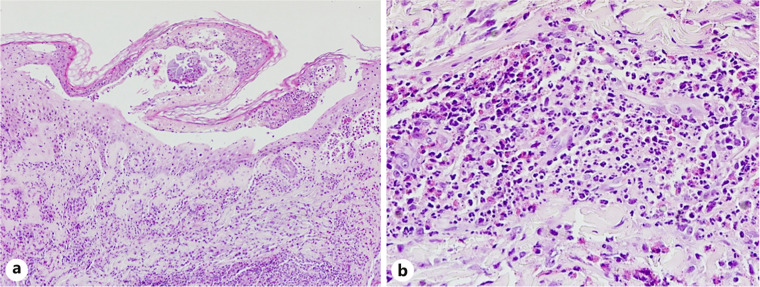
Case presentation: A 40-year-old woman with a history of late-onset allergic rhinitis presented with a 2-week history of numbness in her right leg, along with multiple erythematous papules and vesicles, some with shallow erosions, located on the forehead and left ear. She also experienced fever, progressive dyspnea, and hemoptysis. She was diagnosed with pneumonitis, alveolar hemorrhage, and mononeuritis of the right leg. Laboratory findings revealed leukocytosis with eosinophilia, and the anti-myeloperoxidase antibody was positive. Histopathological examination of the bullous lesion on the forehead showed intraepidermal separation with necrotic keratinocytes and prominent eosinophil infiltration, along with focal leukocytoclastic vasculitis. The patient was diagnosed with EGPA and started on intravenous steroids and cyclophosphamide. EGPA is a rare disease characterized by multiorgan vasculitis, asthma, and granulomatous eosinophilic inflammation, which are its key hallmarks. While cutaneous involvement is common, bullous vasculitis is rarely observed on the forehead and ear.
Conclusions: EGPA is a challenging diagnosis due to its variable presentation. While cutaneous manifestations are common, widespread bullous vasculitis may be atypical and rare clinical presentation. This case underscores the importance of considering EGPA in the differential diagnosis of bullous vasculitis, particularly when associated with systemic symptoms and eosinophilia. Early recognition and treatment are crucial for improving outcomes in this potentially life-threatening condition.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


