Deciphering the phenotypic spectrum associated with MIA3-related odontochondrodysplasia
IF 2.5
3区 生物学
Q2 GENETICS & HEREDITY
引用次数: 0
Abstract
Odontochondrodysplasia (ODCD) is a rare skeletal dysplasia characterized by short stature, skeletal deformities, and dentinogenesis imperfecta (DI). Although the majority of cases were associated with biallelic variants in TRIP11, one study described a homozygous truncating variant in MIA3, encoding TANGO1, in four sibs with ODCD in association with insulin-dependent diabetes, hearing loss, obesity, and intellectual disability. Subsequently, a homozygous truncating variant in the luminal domain of TANGO1 was identified in a fetus with a lethal skeletal dysplasia and fetal hydrops. Herein, we describe two unrelated patients with a distinct phenotype including severe short limbs, short stature, metaphyseal dysplasia, dysmorphic facies, lax joints, and DI. Other variable features were scoliosis, squint, and cardiac problems. Exome sequencing revealed two homozygous MIA3 variants in the luminal domain of TANGO1, c.354+2T>G and p.Cys38Phe. The c.354+2T>G variant was confirmed by investigating the patient’s mRNA to result in exon 3 skipping and an inframe deletion of 29 amino acids. Our patients lacked the extra-skeletal manifestations noted in the four sibs with MIA3 variant. However, they had more severe skeletal deformities closely resembling those observed in patients with TRIP11 variants. Our study suggests the presence of a phenotypic spectrum associated with MIA3 variants including ODCD with milder skeletal deformities, a classic ODCD with severe skeletal deformities, and a lethal skeletal dysplasia at the severe end of the spectrum. Although the striking phenotypic variability appears to be related to the type and or the location of the MIA3 variants, the influence of other factors cannot be ruled out.
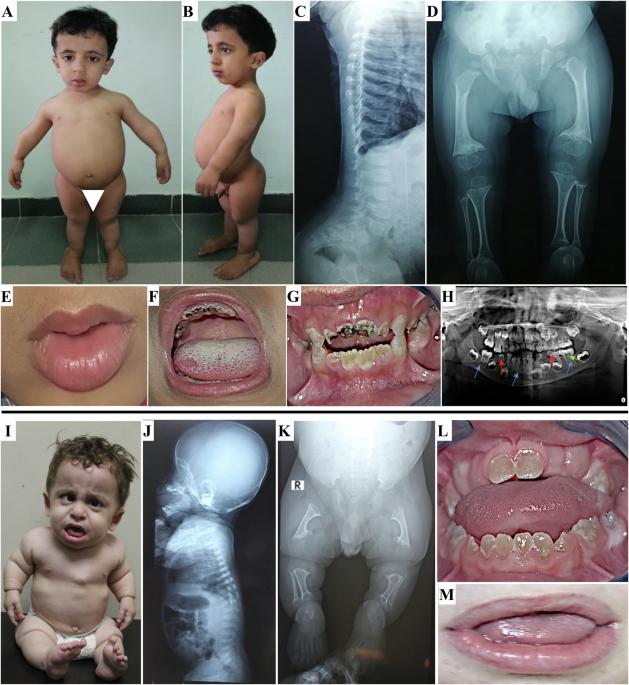
破译与mia3相关的牙软骨发育不良相关的表型谱。
牙髓软骨发育不良(ODCD)是一种罕见的骨骼发育不良,以身材矮小、骨骼畸形和牙本质发育不全(DI)为特征。尽管大多数病例与TRIP11的双等位基因变异有关,但一项研究描述了MIA3的纯合截断变异,编码TANGO1,在患有ODCD的四个兄弟姐妹中与胰岛素依赖型糖尿病、听力损失、肥胖和智力残疾有关。随后,TANGO1的管腔结构域的纯合子截断变异在一个致命的骨骼发育不良和胎儿水肿的胎儿中被发现。在本文中,我们描述了两名不相关的患者,他们具有不同的表型,包括严重的四肢短小、身材矮小、干骺端发育不良、畸形相、关节松弛和DI。其他可变特征包括脊柱侧凸、斜视和心脏问题。外显子组测序显示TANGO1的光腔结构域有两个纯合子MIA3变异,c.354+2T>G和p.Cys38Phe。通过研究患者的mRNA,证实了c.354+2T >g变异导致外显子3跳变和29个氨基酸的框内缺失。我们的患者缺乏MIA3变异的4个兄弟姐妹的骨骼外表现。然而,他们有更严重的骨骼畸形,与在TRIP11变异患者中观察到的非常相似。我们的研究表明,存在与MIA3变异相关的表型谱,包括伴有轻度骨骼畸形的ODCD,伴有严重骨骼畸形的典型ODCD,以及在谱系的严重端出现致命的骨骼发育不良。尽管惊人的表型变异似乎与MIA3变异的类型和/或位置有关,但不能排除其他因素的影响。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Journal of Human Genetics
生物-遗传学
CiteScore
7.20
自引率
0.00%
发文量
101
审稿时长
4-8 weeks
期刊介绍:
The Journal of Human Genetics is an international journal publishing articles on human genetics, including medical genetics and human genome analysis. It covers all aspects of human genetics, including molecular genetics, clinical genetics, behavioral genetics, immunogenetics, pharmacogenomics, population genetics, functional genomics, epigenetics, genetic counseling and gene therapy.
Articles on the following areas are especially welcome: genetic factors of monogenic and complex disorders, genome-wide association studies, genetic epidemiology, cancer genetics, personal genomics, genotype-phenotype relationships and genome diversity.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


