Bridging Oxide Thermodynamics and Site-Blocking: A Computational Study of ORR Activity on Platinum Nanoparticles
IF 13.1
1区 化学
Q1 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
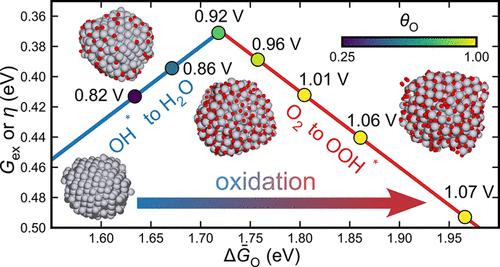
The oxygen reduction reaction (ORR) is a key reaction in fuel cells and metal–air batteries, where high overpotentials remain a critical challenge despite extensive research. While experimental studies have revealed the importance of surface oxidation, a unified computational framework capable of simultaneously capturing both the thermodynamic aspects of rate-determining steps and the kinetic effects of site-blocking on the overpotential has remained elusive. In this work, we present a computational approach that bridges this gap by combining grand-canonical Monte Carlo simulations with the MACE-MP-0 foundation model to study the ORR on experimentally reconstructed Pt nanoparticles. This framework enables the systematic investigation of oxidation effects across multiple scales, from atomic-level place-exchange mechanisms to macroscopic kinetic behavior. Our simulations reveal a strong dependence of system thermodynamics on oxygen coverage and successfully predict the place-exchange mechanism onset at 1.06 V vs SHE, in agreement with experimental observations. Through established scaling relations and deletion energy analysis, we quantify both the rate-determining step and the distribution of reactive sites on the oxidized surface, providing insight into the complex interplay between surface oxidation and ORR activity. By linking our results with both theoretical and experimental benchmarks on multiple points, we ensure the viability of our assumptions and approach. Using a simplified kinetic model derived from our simulations, we demonstrate agreement with core experimental observations, illustrating how computational approaches based on foundation models can enhance our understanding of catalytic processes. This work not only provides a comprehensive understanding of oxide effects in ORR but also establishes a versatile computational methodology that can be readily extended to study similar electrochemical processes on other catalytic systems, offering a powerful tool for rational catalyst design.
桥接氧化物热力学和位点阻断:铂纳米颗粒上ORR活性的计算研究
氧还原反应(ORR)是燃料电池和金属-空气电池中的一个关键反应,尽管在这些领域进行了广泛的研究,但高过电位仍然是一个严峻的挑战。虽然实验研究已经揭示了表面氧化的重要性,但能够同时捕获速率决定步骤的热力学方面和位点阻塞对过电位的动力学影响的统一计算框架仍然难以捉摸。在这项工作中,我们提出了一种计算方法,通过将大规范蒙特卡罗模拟与MACE-MP-0基础模型相结合来研究实验重建Pt纳米粒子的ORR,从而弥补了这一差距。该框架使系统地研究氧化效应跨越多个尺度,从原子水平的位置交换机制到宏观动力学行为。我们的模拟揭示了系统热力学对氧覆盖的强烈依赖,并成功地预测了在1.06 V vs SHE下位置交换机制的开始,与实验观察结果一致。通过建立标度关系和缺失能分析,我们量化了速率决定步骤和氧化表面上反应位点的分布,从而深入了解表面氧化与ORR活性之间的复杂相互作用。通过将我们的结果与多个点的理论和实验基准联系起来,我们确保了我们的假设和方法的可行性。使用从我们的模拟中得到的简化动力学模型,我们证明了与核心实验观察的一致性,说明了基于基础模型的计算方法如何增强我们对催化过程的理解。这项工作不仅提供了对ORR中氧化物效应的全面理解,而且建立了一种通用的计算方法,可以很容易地扩展到研究其他催化系统中类似的电化学过程,为合理设计催化剂提供了有力的工具。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

ACS Catalysis
CHEMISTRY, PHYSICAL-
CiteScore
20.80
自引率
6.20%
发文量
1253
审稿时长
1.5 months
期刊介绍:
ACS Catalysis is an esteemed journal that publishes original research in the fields of heterogeneous catalysis, molecular catalysis, and biocatalysis. It offers broad coverage across diverse areas such as life sciences, organometallics and synthesis, photochemistry and electrochemistry, drug discovery and synthesis, materials science, environmental protection, polymer discovery and synthesis, and energy and fuels.
The scope of the journal is to showcase innovative work in various aspects of catalysis. This includes new reactions and novel synthetic approaches utilizing known catalysts, the discovery or modification of new catalysts, elucidation of catalytic mechanisms through cutting-edge investigations, practical enhancements of existing processes, as well as conceptual advances in the field. Contributions to ACS Catalysis can encompass both experimental and theoretical research focused on catalytic molecules, macromolecules, and materials that exhibit catalytic turnover.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


