{"title":"Zinner syndrome: report of a case and whole exome sequencing.","authors":"Jiatai He, Chengcheng Wei, Yu Huang, Feixiang Xu, Miao Wang, Zhaohui Chen","doi":"10.1186/s12610-025-00256-3","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Zinner syndrome is a rare congenital malformation of the male genitourinary system, characterized by a triad: seminal vesicle cyst, unilateral renal agenesis, and ipsilateral ejaculatory duct obstruction. The etiology of this uncommon disease remains largely elusive; however, genetic mutations may contribute to its development. In this report, we present a case of symptomatic Zinner syndrome that was surgically treated, alongside an investigation into the potential genetic basis of the syndrome via whole exome sequencing.</p><p><strong>Case presentation: </strong>We report the case of an 18-year-old male presenting with urinary pain and was diagnosed with right renal agenesis and a left seminal vesicle cyst following comprehensive imaging. The patient also experienced perineal pain and urgency, without symptoms of frequent urination, dysuria, or hematuria, and no familial history of genitourinary anomalies was documented. He successfully underwent laparoscopic resection of a pelvic mass, with pathological examination confirming a seminal vesicle cyst. Postoperative recovery was uneventful. Whole exome sequencing of blood and tissue samples highlighted myeloma overexpressed gene (MYEOV), B melanoma antigen family member (BAGE), and N-acetylated-alpha-linked acidic dipeptidase 2 (NAALAD2) as potential mutated genes related to Zinner syndrome. Additionally, two predisposing genetic variants were identified.</p><p><strong>Conclusions: </strong>Zinner syndrome is a rare condition commonly diagnosed via various imaging modalities. Surgical resection remains the most effective treatment for symptomatic cases. Gene sequencing provides valuable insights into the genetic etiology of Zinner syndrome, enhancing our understanding and potentially guiding future diagnostic approaches.</p>","PeriodicalId":8730,"journal":{"name":"Basic and Clinical Andrology","volume":"35 1","pages":"10"},"PeriodicalIF":2.0000,"publicationDate":"2025-03-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11895205/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Basic and Clinical Andrology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s12610-025-00256-3","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"ANDROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Zinner syndrome is a rare congenital malformation of the male genitourinary system, characterized by a triad: seminal vesicle cyst, unilateral renal agenesis, and ipsilateral ejaculatory duct obstruction. The etiology of this uncommon disease remains largely elusive; however, genetic mutations may contribute to its development. In this report, we present a case of symptomatic Zinner syndrome that was surgically treated, alongside an investigation into the potential genetic basis of the syndrome via whole exome sequencing.
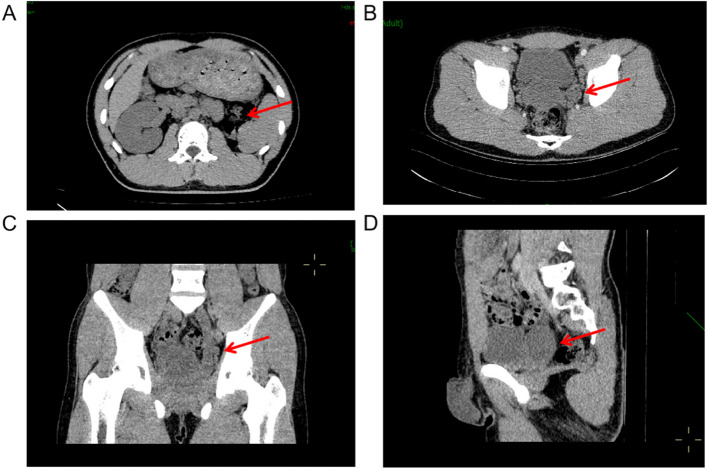
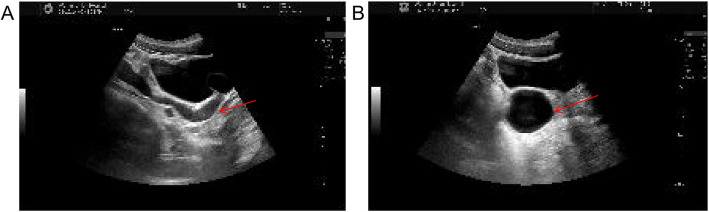
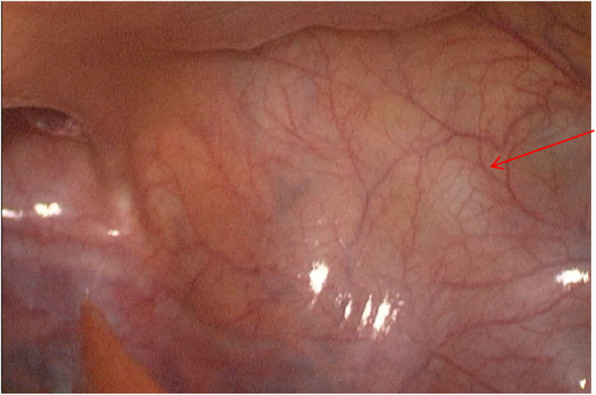
Case presentation: We report the case of an 18-year-old male presenting with urinary pain and was diagnosed with right renal agenesis and a left seminal vesicle cyst following comprehensive imaging. The patient also experienced perineal pain and urgency, without symptoms of frequent urination, dysuria, or hematuria, and no familial history of genitourinary anomalies was documented. He successfully underwent laparoscopic resection of a pelvic mass, with pathological examination confirming a seminal vesicle cyst. Postoperative recovery was uneventful. Whole exome sequencing of blood and tissue samples highlighted myeloma overexpressed gene (MYEOV), B melanoma antigen family member (BAGE), and N-acetylated-alpha-linked acidic dipeptidase 2 (NAALAD2) as potential mutated genes related to Zinner syndrome. Additionally, two predisposing genetic variants were identified.
Conclusions: Zinner syndrome is a rare condition commonly diagnosed via various imaging modalities. Surgical resection remains the most effective treatment for symptomatic cases. Gene sequencing provides valuable insights into the genetic etiology of Zinner syndrome, enhancing our understanding and potentially guiding future diagnostic approaches.
期刊介绍:
Basic and Clinical Andrology is an open access journal in the domain of andrology covering all aspects of male reproductive and sexual health in both human and animal models. The journal aims to bring to light the various clinical advancements and research developments in andrology from the international community.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


