Automated model-free analysis of cryo-EM volume ensembles with SIREn
IF 4.4
2区 生物学
Q2 BIOCHEMISTRY & MOLECULAR BIOLOGY
引用次数: 0
Abstract
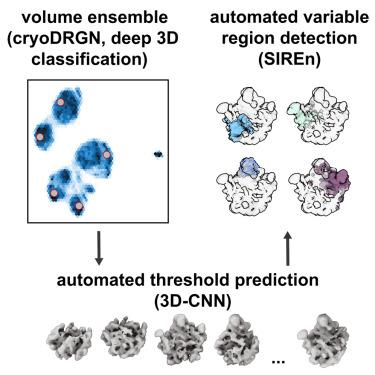
Cryogenic electron microscopy (cryo-EM) has the potential to capture snapshots of proteins in motion and generate hypotheses linking conformational states to biological function. This potential has been increasingly realized by the advent of machine learning models that allow 100s-1,000s of 3D density maps to be generated from a single dataset. How to identify distinct structural states within these volume ensembles and quantify their relative occupancies remain open questions. Here, we present an approach to inferring variable regions directly from a volume ensemble based on the statistical co-occupancy of voxels, as well as a 3D convolutional neural network that predicts binarization thresholds for volumes in an unbiased and automated manner. We show that these tools recapitulate known heterogeneity in a variety of simulated and real cryo-EM datasets and highlight how integrating these tools with existing data processing pipelines enables improved particle curation.
求助全文
约1分钟内获得全文
求助全文
来源期刊

Structure
生物-生化与分子生物学
CiteScore
8.90
自引率
1.80%
发文量
155
审稿时长
3-8 weeks
期刊介绍:
Structure aims to publish papers of exceptional interest in the field of structural biology. The journal strives to be essential reading for structural biologists, as well as biologists and biochemists that are interested in macromolecular structure and function. Structure strongly encourages the submission of manuscripts that present structural and molecular insights into biological function and mechanism. Other reports that address fundamental questions in structural biology, such as structure-based examinations of protein evolution, folding, and/or design, will also be considered. We will consider the application of any method, experimental or computational, at high or low resolution, to conduct structural investigations, as long as the method is appropriate for the biological, functional, and mechanistic question(s) being addressed. Likewise, reports describing single-molecule analysis of biological mechanisms are welcome.
In general, the editors encourage submission of experimental structural studies that are enriched by an analysis of structure-activity relationships and will not consider studies that solely report structural information unless the structure or analysis is of exceptional and broad interest. Studies reporting only homology models, de novo models, or molecular dynamics simulations are also discouraged unless the models are informed by or validated by novel experimental data; rationalization of a large body of existing experimental evidence and making testable predictions based on a model or simulation is often not considered sufficient.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


