Mingchuang He, Rongxing Zhang, Tongkun Wang, Xiao-Song Xue, Dawei Ma
{"title":"Assembly of (hetero)aryl sulfilimines via copper-catalyzed enantioselective S-arylation of sulfenamides with (hetero)aryl Iodides","authors":"Mingchuang He, Rongxing Zhang, Tongkun Wang, Xiao-Song Xue, Dawei Ma","doi":"10.1038/s41467-025-57474-6","DOIUrl":null,"url":null,"abstract":"<p>The (hetero)aryl sulfoximines are important structures for developing bioactive molecules, whose synthesis relies on oxidation of (hetero)aryl sulfilimines. However, asymmetric approaches for assembling (hetero)aryl sulfilimines are still rare. Here we show that combination of CuI and NOBIN-derived amide ligands offers an effective catalytic system for enantioselective coupling of (hetero)aryl iodides with sulfenamides. A large number of functional groups and heterocycles are tolerated under the coupling conditions, providing a powerful approach for diverse synthesis of enantioenriched (hetero)aryl sulfilimines. The efficiency of the coupling reaction is highly dependent on the electronic nature of (hetero)aryl iodides and sulfenamides. Both (hetero)aryl- and some bulky alkyl-substituted sulfenamides give excellent enantioselectivities, while sulfenamides with smaller alkyl substituents lead to the formation of the (hetero)aryl sulfilimines with moderate enantioselectivities. Density functional theory (DFT) calculations reveal that proper steric repulsions in the transition states of the intramolecular S<sub>N</sub>Ar reaction are crucial for achieving desirable enantioselectivity.</p>","PeriodicalId":19066,"journal":{"name":"Nature Communications","volume":"12 1","pages":""},"PeriodicalIF":14.7000,"publicationDate":"2025-03-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nature Communications","FirstCategoryId":"103","ListUrlMain":"https://doi.org/10.1038/s41467-025-57474-6","RegionNum":1,"RegionCategory":"综合性期刊","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MULTIDISCIPLINARY SCIENCES","Score":null,"Total":0}
引用次数: 0
Abstract
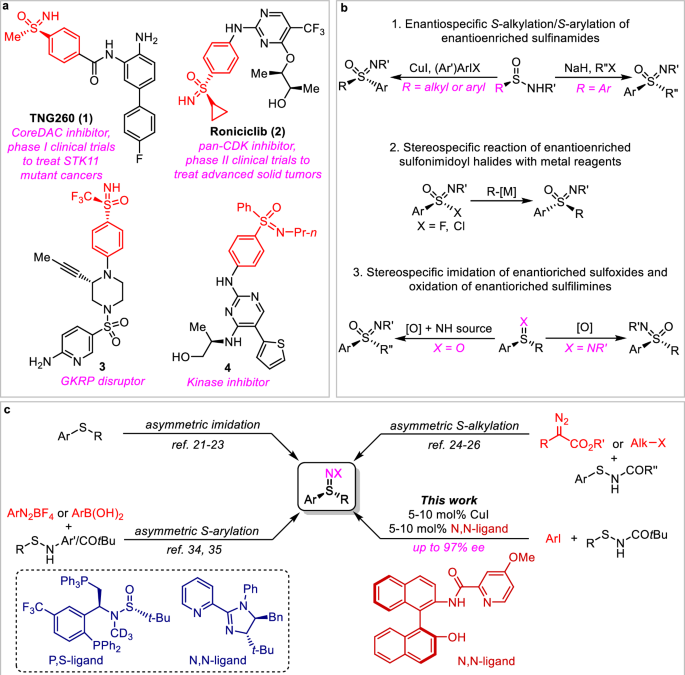
The (hetero)aryl sulfoximines are important structures for developing bioactive molecules, whose synthesis relies on oxidation of (hetero)aryl sulfilimines. However, asymmetric approaches for assembling (hetero)aryl sulfilimines are still rare. Here we show that combination of CuI and NOBIN-derived amide ligands offers an effective catalytic system for enantioselective coupling of (hetero)aryl iodides with sulfenamides. A large number of functional groups and heterocycles are tolerated under the coupling conditions, providing a powerful approach for diverse synthesis of enantioenriched (hetero)aryl sulfilimines. The efficiency of the coupling reaction is highly dependent on the electronic nature of (hetero)aryl iodides and sulfenamides. Both (hetero)aryl- and some bulky alkyl-substituted sulfenamides give excellent enantioselectivities, while sulfenamides with smaller alkyl substituents lead to the formation of the (hetero)aryl sulfilimines with moderate enantioselectivities. Density functional theory (DFT) calculations reveal that proper steric repulsions in the transition states of the intramolecular SNAr reaction are crucial for achieving desirable enantioselectivity.
期刊介绍:
Nature Communications, an open-access journal, publishes high-quality research spanning all areas of the natural sciences. Papers featured in the journal showcase significant advances relevant to specialists in each respective field. With a 2-year impact factor of 16.6 (2022) and a median time of 8 days from submission to the first editorial decision, Nature Communications is committed to rapid dissemination of research findings. As a multidisciplinary journal, it welcomes contributions from biological, health, physical, chemical, Earth, social, mathematical, applied, and engineering sciences, aiming to highlight important breakthroughs within each domain.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


