Catalytic molten Zn-Bi alloys for methane pyrolysis
IF 13.3
1区 工程技术
Q1 ENGINEERING, CHEMICAL
引用次数: 0
Abstract
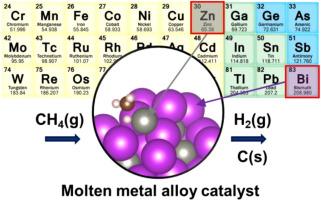
Methane pyrolysis with molten alloy catalysts enables the production of large-scale, CO2-free hydrogen and valuable carbon byproducts. This work systematically screens molten alloy catalysts, determines reaction kinetics, elucidates detailed surface reaction mechanisms, and analyzes the structure of carbon byproducts using both computational and experimental methods. Several essential factors for designing Bi-based molten alloys suggest that Zn0.45-Bi0.55 is a promising candidate among 20 binary alloys. We calculate the accurate free energy of activation for the initial C-H activation of methane using ab initio molecular dynamics and metadynamics simulations. The computed barrier is lower than those of molten binary alloys reported in the literature, and this has been validated by our reaction kinetics measurements on the Zn-Bi alloy. In methane activation, active metals (Zn) contribute to changing the charge states of base metals (Bi), facilitating C-H dissociation. Methane activation is more likely to occur through a surface-stabilized-like pathway rather than a radical-initiated pathway, which could provide crucial information for developing a descriptor to predict C-H activation energies on molten catalysts. A distinct feature of the surface-stabilized-like pathway, compared to solid surfaces, is that methyl does not necessarily bind to the active site immediately after C-H dissociation. We also investigate methane decomposition and carbon formation pathways using density functional theory calculations. The initial C-C bond can form either through CH3(g) radicals in the gas phase or via coupling reactions involving CH2* and CH*. Transmission electron microscopy of the carbon products shows a partially crystalline structure, suggesting their potential usage as high-value carbon.
求助全文
约1分钟内获得全文
求助全文
来源期刊

Chemical Engineering Journal
工程技术-工程:化工
CiteScore
21.70
自引率
9.30%
发文量
6781
审稿时长
2.4 months
期刊介绍:
The Chemical Engineering Journal is an international research journal that invites contributions of original and novel fundamental research. It aims to provide an international platform for presenting original fundamental research, interpretative reviews, and discussions on new developments in chemical engineering. The journal welcomes papers that describe novel theory and its practical application, as well as those that demonstrate the transfer of techniques from other disciplines. It also welcomes reports on carefully conducted experimental work that is soundly interpreted. The main focus of the journal is on original and rigorous research results that have broad significance. The Catalysis section within the Chemical Engineering Journal focuses specifically on Experimental and Theoretical studies in the fields of heterogeneous catalysis, molecular catalysis, and biocatalysis. These studies have industrial impact on various sectors such as chemicals, energy, materials, foods, healthcare, and environmental protection.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


