{"title":"Nonadiabatic Coupling Dictates the Site-Specific Excited-State Decay Pathways of Fluorophenols.","authors":"Jayshree Sadhukhan, Moitrayee Mukherjee, Piyali Chatterjee, Anwesha Datta","doi":"10.1021/acsomega.4c11321","DOIUrl":null,"url":null,"abstract":"<p><p>In this paper, a combined photophysical and electronic structure theory study demonstrating a remarkable site-specific fluorine substitution effect on the excited-state dynamics of monofluorophenols has been presented. The S<sub>1</sub> ← S<sub>0</sub> electronic origin band of phenol is shifted to a longer wavelength for <i>para</i> substitution, but to shorter wavelengths for <i>ortho</i> and <i>meta</i> substitutions. The observed sequence of excitation wavelengths of 2-fluorophenol (2FP) < 3-fluorophenol (3FP) < phenol < 4-fluorophenol (4FP) is consistent with the transition energies predicted by TDDFT/CAMB3LYP/6-311++G(d,p) and CASSCF(8,8)/Dunning cc-pVDZ theoretical methods. The most notable contrast of excited-state dynamics is revealed in the different features of the fluorescence spectra; the fluorescence yield of 4FP is almost 6 times larger compared to that of 3FP and the spectral bandwidth of 2FP is nearly 1.5 times larger than that of 4FP. Electronic structure calculation predicts a low-energy S<sub>1</sub>/S<sub>0</sub> conical intersection (CI) near the <sup>1</sup>ππ* minimum with respect to the prefulvenic vibronic mode of the aromatic ring, and the energetic location of this CI is altered with the substitution site of the fluorine atom. The predicted energy barrier to this prefulvenic CI is smallest for 3FP but largest for 4FP, leading to a facilitated nonradiative electronic relaxation of the former (3FP), and emission occurs with a much diminished fluorescence intensity.</p>","PeriodicalId":22,"journal":{"name":"ACS Omega","volume":"10 7","pages":"7389-7399"},"PeriodicalIF":4.3000,"publicationDate":"2025-02-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11866181/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"ACS Omega","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acsomega.4c11321","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/2/25 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
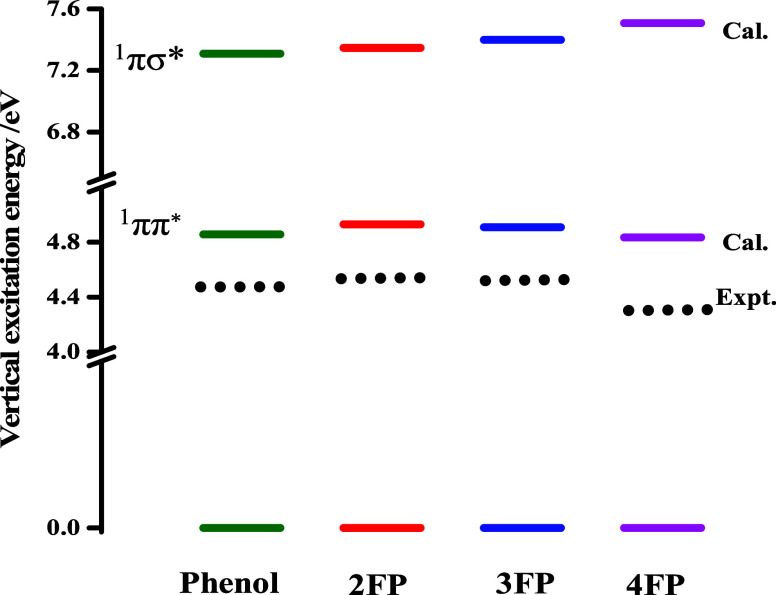
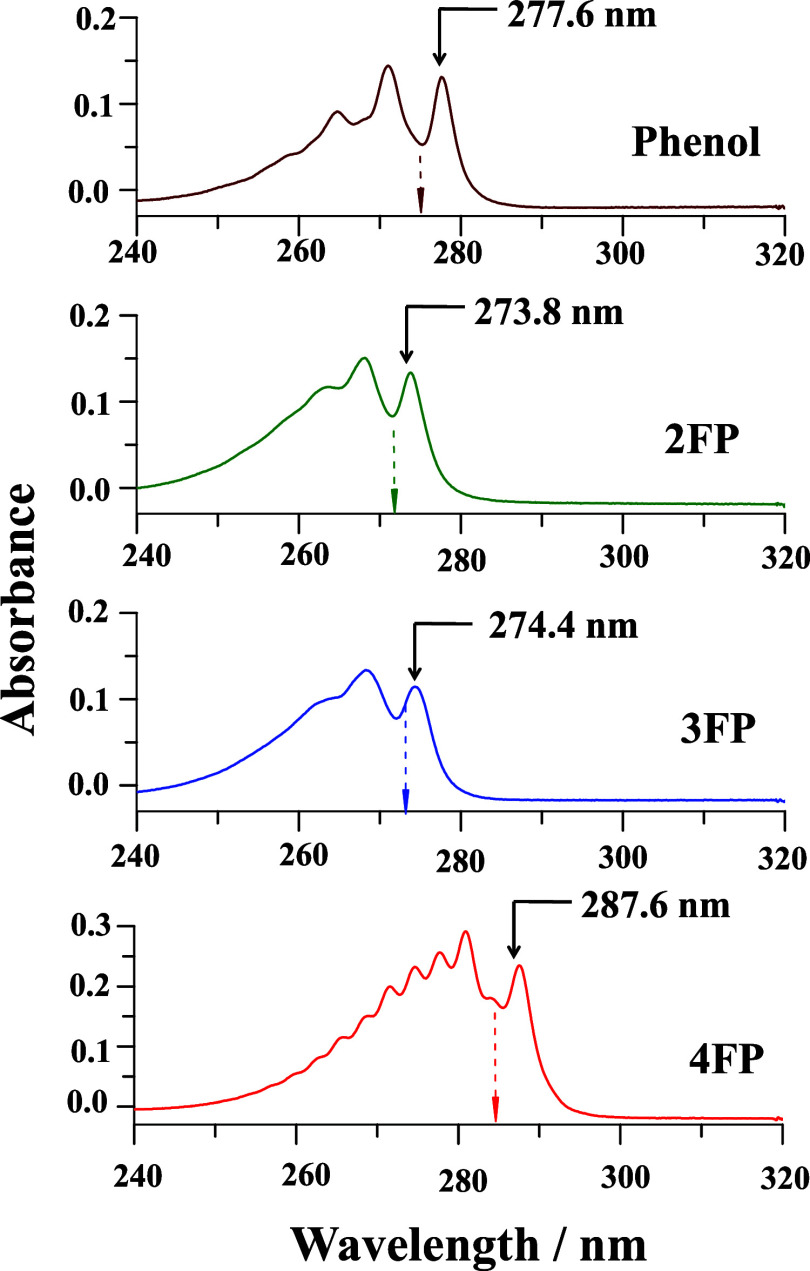
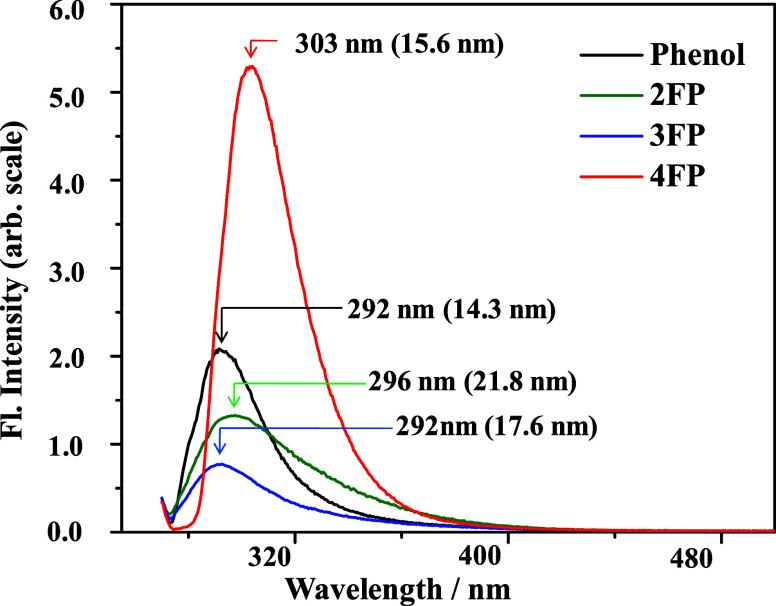
In this paper, a combined photophysical and electronic structure theory study demonstrating a remarkable site-specific fluorine substitution effect on the excited-state dynamics of monofluorophenols has been presented. The S1 ← S0 electronic origin band of phenol is shifted to a longer wavelength for para substitution, but to shorter wavelengths for ortho and meta substitutions. The observed sequence of excitation wavelengths of 2-fluorophenol (2FP) < 3-fluorophenol (3FP) < phenol < 4-fluorophenol (4FP) is consistent with the transition energies predicted by TDDFT/CAMB3LYP/6-311++G(d,p) and CASSCF(8,8)/Dunning cc-pVDZ theoretical methods. The most notable contrast of excited-state dynamics is revealed in the different features of the fluorescence spectra; the fluorescence yield of 4FP is almost 6 times larger compared to that of 3FP and the spectral bandwidth of 2FP is nearly 1.5 times larger than that of 4FP. Electronic structure calculation predicts a low-energy S1/S0 conical intersection (CI) near the 1ππ* minimum with respect to the prefulvenic vibronic mode of the aromatic ring, and the energetic location of this CI is altered with the substitution site of the fluorine atom. The predicted energy barrier to this prefulvenic CI is smallest for 3FP but largest for 4FP, leading to a facilitated nonradiative electronic relaxation of the former (3FP), and emission occurs with a much diminished fluorescence intensity.
ACS OmegaChemical Engineering-General Chemical Engineering
CiteScore
6.60
自引率
4.90%
发文量
3945
审稿时长
2.4 months
期刊介绍:
ACS Omega is an open-access global publication for scientific articles that describe new findings in chemistry and interfacing areas of science, without any perceived evaluation of immediate impact.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


