{"title":"Novel <i>SPAST</i> Deletion Mutation in an American Family With Hereditary Spastic Paraplegia: A Case Report.","authors":"Sydney B Bhopatkar, Juebin Huang","doi":"10.1177/23247096251323173","DOIUrl":null,"url":null,"abstract":"<p><p>The diverse group of neurodegenerative disorders known as hereditary spastic paraplegia (HSP) is characterized by spasticity and weakness of the bilateral lower extremity due to degeneration of the corticospinal tract. The pathogenesis of HSP is broad, with autosomal dominant, autosomal recessive, X-linked recessive, mitochondrial inheritance, and de novo mutations reported, along with remarkable heterogeneity of mutations and clinical presentation. Of these, the most common subtype of HSP is HSP type 4 (HSP-<i>SPG4</i>), a result of mutations in the <i>SPAST</i> gene (chromosome 2p22.3) that leads to impaired activity of the microtubule-severing protein spastin. Typically presenting as an uncomplicated, autosomal dominant form of the disease, HSP-<i>SPG4</i> has been documented worldwide with vast genomic variance across the <i>SPAST</i> gene. Despite common features in clinical phenotypes, a clear link between <i>SPAST</i> gene variants and disease presentation remains vague. Here, we report a novel 26.1 kb deletion in the <i>SPAST</i> gene (del exons 4-7) in a US family with previously undiagnosed HSP-<i>SPG4</i>.</p>","PeriodicalId":16198,"journal":{"name":"Journal of investigative medicine high impact case reports","volume":"13 ","pages":"23247096251323173"},"PeriodicalIF":0.8000,"publicationDate":"2025-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11869264/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of investigative medicine high impact case reports","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1177/23247096251323173","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"MEDICINE, GENERAL & INTERNAL","Score":null,"Total":0}
引用次数: 0
Abstract
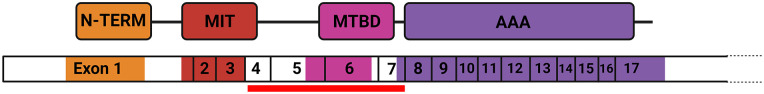
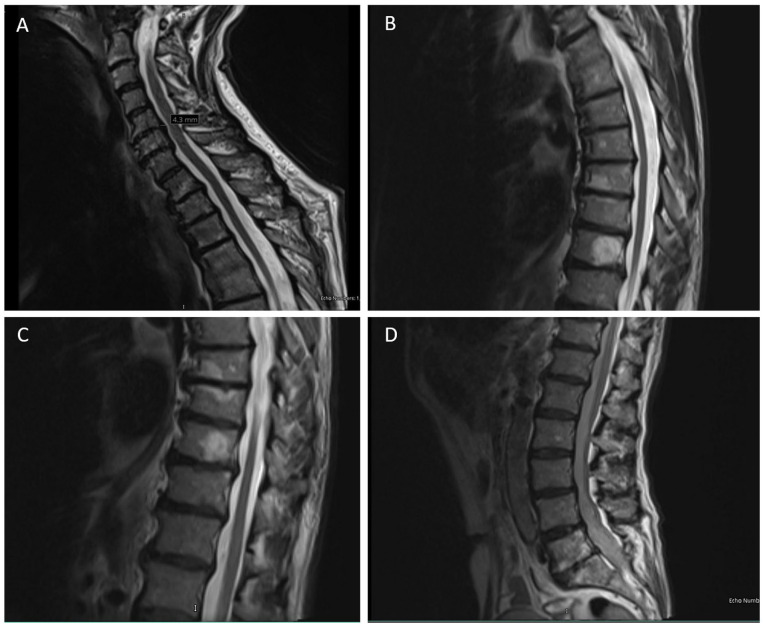
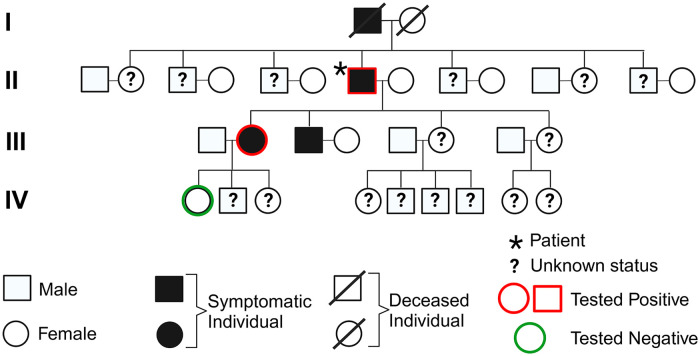
The diverse group of neurodegenerative disorders known as hereditary spastic paraplegia (HSP) is characterized by spasticity and weakness of the bilateral lower extremity due to degeneration of the corticospinal tract. The pathogenesis of HSP is broad, with autosomal dominant, autosomal recessive, X-linked recessive, mitochondrial inheritance, and de novo mutations reported, along with remarkable heterogeneity of mutations and clinical presentation. Of these, the most common subtype of HSP is HSP type 4 (HSP-SPG4), a result of mutations in the SPAST gene (chromosome 2p22.3) that leads to impaired activity of the microtubule-severing protein spastin. Typically presenting as an uncomplicated, autosomal dominant form of the disease, HSP-SPG4 has been documented worldwide with vast genomic variance across the SPAST gene. Despite common features in clinical phenotypes, a clear link between SPAST gene variants and disease presentation remains vague. Here, we report a novel 26.1 kb deletion in the SPAST gene (del exons 4-7) in a US family with previously undiagnosed HSP-SPG4.
期刊介绍:
The AFMR is committed to enhancing the training and career development of our members and to furthering its mission to facilitate the conduct of research to improve medical care. Case reports represent an important avenue for trainees (interns, residents, and fellows) and early-stage faculty to demonstrate productive, scholarly activity.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


