Elizabeth Carbonell, Sarah L Stenton, Vijay S Ganesh, Jialan Ma, Grace E VanNoy, Lynn Pais, John N Gaitanis, Melanie C O'Leary, Heidi L Rehm, Anne O'Donnell-Luria
{"title":"Male proband with intractable seizures and a de novo start-codon-disrupting variant in GLUL.","authors":"Elizabeth Carbonell, Sarah L Stenton, Vijay S Ganesh, Jialan Ma, Grace E VanNoy, Lynn Pais, John N Gaitanis, Melanie C O'Leary, Heidi L Rehm, Anne O'Donnell-Luria","doi":"10.1016/j.xhgg.2025.100419","DOIUrl":null,"url":null,"abstract":"<p><p>Bi-allelic variants in GLUL, encoding glutamine synthetase and responsible for the conversion of glutamate to glutamine, are associated with a severe recessive disease due to glutamine deficiency. A dominant disease mechanism was recently reported in nine females, all with a de novo single-nucleotide variant within the start codon or the 5' UTR of GLUL that truncates 17 amino acids of the protein product, including its critical N-terminal degron sequence. This truncation results in a disorder of abnormal glutamine synthetase stability and manifests as a phenotype of severe developmental and epileptic encephalopathy. Here, we report the first male with a pathogenic de novo variant in the same critical region of GLUL, with a phenotype of refractory focal and generalized seizures, as well as developmental delays. We provide a detailed description of the disease course and treatment response.</p>","PeriodicalId":34530,"journal":{"name":"HGG Advances","volume":" ","pages":"100419"},"PeriodicalIF":3.6000,"publicationDate":"2025-04-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11930682/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"HGG Advances","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1016/j.xhgg.2025.100419","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/2/21 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
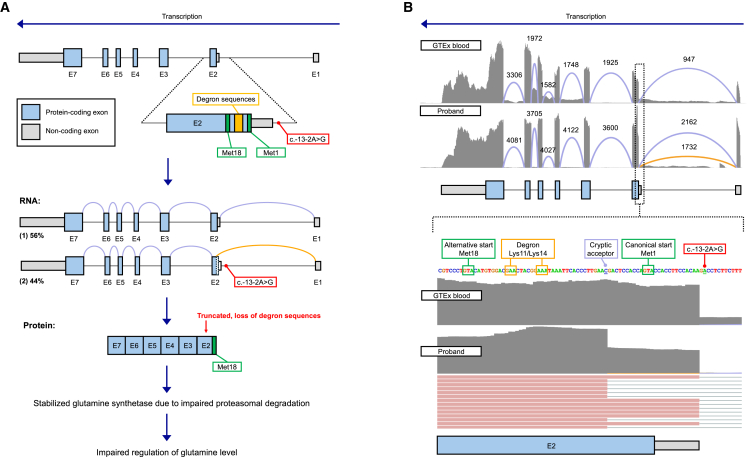
Bi-allelic variants in GLUL, encoding glutamine synthetase and responsible for the conversion of glutamate to glutamine, are associated with a severe recessive disease due to glutamine deficiency. A dominant disease mechanism was recently reported in nine females, all with a de novo single-nucleotide variant within the start codon or the 5' UTR of GLUL that truncates 17 amino acids of the protein product, including its critical N-terminal degron sequence. This truncation results in a disorder of abnormal glutamine synthetase stability and manifests as a phenotype of severe developmental and epileptic encephalopathy. Here, we report the first male with a pathogenic de novo variant in the same critical region of GLUL, with a phenotype of refractory focal and generalized seizures, as well as developmental delays. We provide a detailed description of the disease course and treatment response.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


