Predicting Reduction Potentials of Blue Copper Proteins Using Quantum Mechanical Calculations
IF 4.3
2区 化学
Q1 CHEMISTRY, INORGANIC & NUCLEAR
引用次数: 0
Abstract
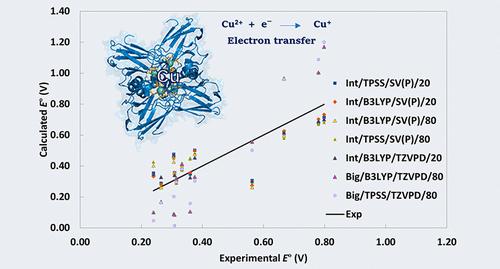
We have calculated redox potentials of 12 blue copper protein sites comparing 64 computational methods, systematically varying the quantum mechanics (QM) system size, dielectric constants, density functional, and basis sets. All methods were based on structures optimized with combined QM and molecular mechanics (QM/MM) approaches. The redox potentials were evaluated using 10 quality metrics. The best results for relative potentials were achieved using a QM system of intermediate size (∼70 atoms), the TPSS density functional, and a SV(P) basis set, using QM-cluster calculations in a continuum solvent with a dielectric constant of 20, yielding a mean absolute deviation of 0.09 V and a maximum deviation of 0.26 V. For absolute redox potentials, methods using larger QM systems (∼340 atoms), the B3LYP density functional, and larger basis sets perform better, achieving mean signed errors down to −0.27 V. Compared to previous studies on iron–sulfur clusters, redox potentials for blue copper proteins show improved accuracy due to their narrower potential range and simpler coordination environments, but systematic errors are system-dependent. This study underscores the challenges of modeling redox-active sites in proteins and highlights the effectiveness of QM-cluster calculations in a continuum solvent in balancing computational cost with predictive power.
求助全文
约1分钟内获得全文
求助全文
来源期刊

Inorganic Chemistry
化学-无机化学与核化学
CiteScore
7.60
自引率
13.00%
发文量
1960
审稿时长
1.9 months
期刊介绍:
Inorganic Chemistry publishes fundamental studies in all phases of inorganic chemistry. Coverage includes experimental and theoretical reports on quantitative studies of structure and thermodynamics, kinetics, mechanisms of inorganic reactions, bioinorganic chemistry, and relevant aspects of organometallic chemistry, solid-state phenomena, and chemical bonding theory. Emphasis is placed on the synthesis, structure, thermodynamics, reactivity, spectroscopy, and bonding properties of significant new and known compounds.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


