Rijad Konjhodzic, Lana Salihefendic, Naida Mulahuseinovic, Ivana Ceko, Selma Durgut, Nejira Handzic, Sadzida Orucevic, Sajra Uzicanin
{"title":"Novel Intronic Heterozygous Mutation in TSC2 Gene in Pediatric Patient with Tuberous Sclerosis Complex.","authors":"Rijad Konjhodzic, Lana Salihefendic, Naida Mulahuseinovic, Ivana Ceko, Selma Durgut, Nejira Handzic, Sadzida Orucevic, Sajra Uzicanin","doi":"10.5455/aim.2024.32.122-125","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Tuberous Sclerosis Complex (TSC) is an autosomal dominant genetic disorder and involves multiple organs, intellectual disability and epilepsy. Mutations in TSC1 and TSC2 genes are responsible for the molecular disease mechanism.</p><p><strong>Objective: </strong>The aim is to determine molecular background of a patient with a suspicion of TSC.</p><p><strong>Case presentation: </strong>In this case report, we describe a seven year old patient with the clinical manifestation of TSC that includes supratentorial changes, subependymal hamartomas and angifibromas in the facial area. Besides the brain and skin changes, no other TSC characteristics were observed. The patient was referred to molecular genetic testing using Next Generation Sequencing (NGS). Results: Clinical exome sequencing revealed intronic TSC2 c.4849+2T>G variant. The variant was confirmed using Sanger sequencing on the subject. However, the variant was not detected in the parents, which indicated that it arose de-novo. The RegSNP-intron, Mutation Taster and Human Splicing Finder were used as a bioinformatic tools to predict the possible effect on protein. Using bioinformatic tools, it was determined that the variant is possibly damaging to protein.</p><p><strong>Conclusion: </strong>This data suggest that observed splicing intronic variant could be the cause of TSC in this pediatric patient.</p>","PeriodicalId":7074,"journal":{"name":"Acta Informatica Medica","volume":"32 2","pages":"122-125"},"PeriodicalIF":0.0000,"publicationDate":"2024-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11821562/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Acta Informatica Medica","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.5455/aim.2024.32.122-125","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"Medicine","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Tuberous Sclerosis Complex (TSC) is an autosomal dominant genetic disorder and involves multiple organs, intellectual disability and epilepsy. Mutations in TSC1 and TSC2 genes are responsible for the molecular disease mechanism.
Objective: The aim is to determine molecular background of a patient with a suspicion of TSC.
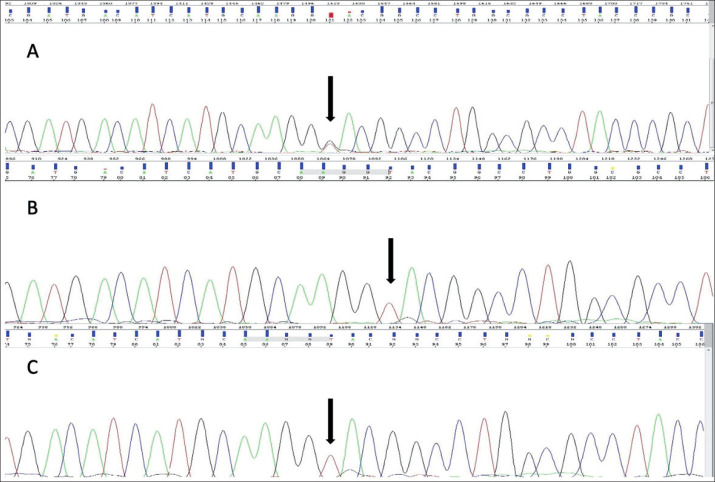
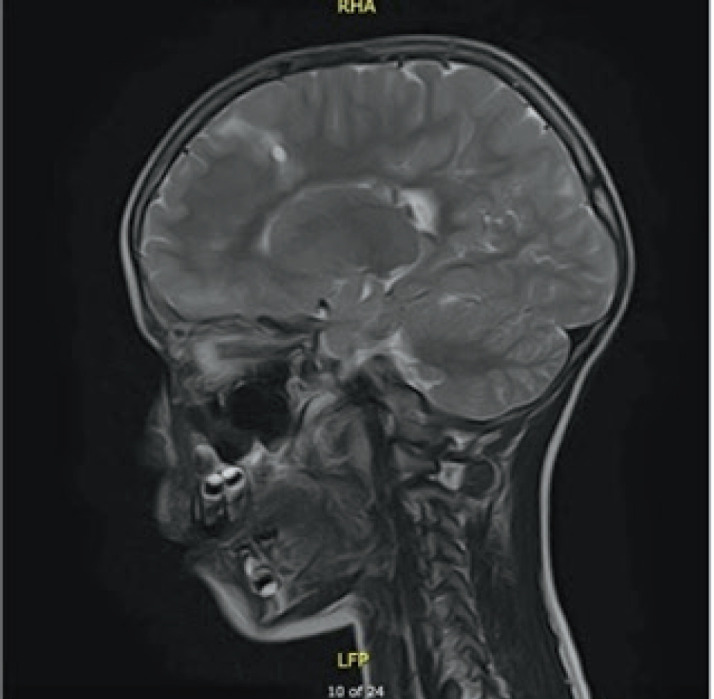
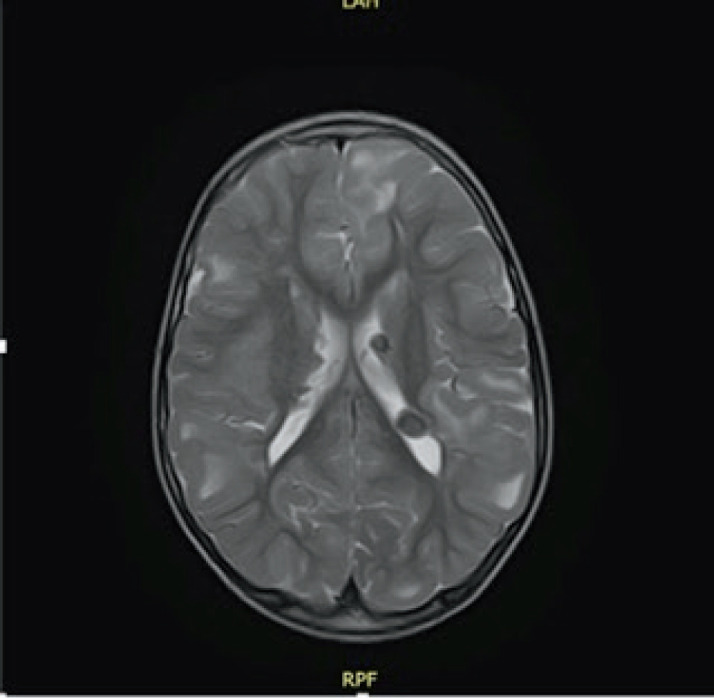
Case presentation: In this case report, we describe a seven year old patient with the clinical manifestation of TSC that includes supratentorial changes, subependymal hamartomas and angifibromas in the facial area. Besides the brain and skin changes, no other TSC characteristics were observed. The patient was referred to molecular genetic testing using Next Generation Sequencing (NGS). Results: Clinical exome sequencing revealed intronic TSC2 c.4849+2T>G variant. The variant was confirmed using Sanger sequencing on the subject. However, the variant was not detected in the parents, which indicated that it arose de-novo. The RegSNP-intron, Mutation Taster and Human Splicing Finder were used as a bioinformatic tools to predict the possible effect on protein. Using bioinformatic tools, it was determined that the variant is possibly damaging to protein.
Conclusion: This data suggest that observed splicing intronic variant could be the cause of TSC in this pediatric patient.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


