Jingyi Li, Gang Zhou, Te Chen, Qiao Lin, Qiupeng Yang
{"title":"LncRNA RMRP promotes chondrocyte injury by regulating the FOXC1/RBP4 axis.","authors":"Jingyi Li, Gang Zhou, Te Chen, Qiao Lin, Qiupeng Yang","doi":"10.5114/ceji.2024.145312","DOIUrl":null,"url":null,"abstract":"<p><strong>Introduction: </strong>The main pathological feature of osteoarthritis (OA) is chondrocyte injury. LncRNA mitochondrial RNA processing endoribonuclease (RMRP) has been shown to be a chondrogenic differentiation factor. This study aimed to explore the role of RMRP in chondrocyte injury.</p><p><strong>Material and methods: </strong>Cell counting kit-8 (CCK-8) and TUNEL assays were used to determine lipopolysaccharide (LPS)-induced chondrocyte viability and apoptosis, respectively. The interaction between RMRP and FOXC1 was analyzed by RIP and RNA pull-down. Dual luciferase reporter and ChIP were employed to analyze the interaction between FOXC1 and RBP4. The levels of RMRP, FOXC1, RBP4, apoptosis-related and extracellular matrix (ECM)-related genes were detected by RT-qPCR and western blot. ELISA assay was used for detection of inflammatory cytokines in LPS-induced chondrocytes.</p><p><strong>Results: </strong>The levels of RMRP, FOXC1 and RBP4 were significantly upregulated in OA cartilage tissues and LPS-induced chondrocytes. Knockdown of RMRP inhibited chondrocyte apoptosis and inflammation under LPS. RMRP interacted with FOXC1 and promoted RBP4 expression. FOXC1 could upregulate RBP4 and promote LPS-induced chondrocyte apoptosis and inflammation. Similarly, RMRP combined with FOXC1 and aggravated apoptosis and inflammation in LPS-treated chondrocytes.</p><p><strong>Conclusions: </strong>RMRP promoted upregulation of RBP4 and activation of the JNK signaling pathway by binding to FOXC1, thereby accelerating LPS-induced apoptosis and inflammation in chondrocytes.</p>","PeriodicalId":9694,"journal":{"name":"Central European Journal of Immunology","volume":"49 4","pages":"366-382"},"PeriodicalIF":1.6000,"publicationDate":"2024-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11811726/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Central European Journal of Immunology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.5114/ceji.2024.145312","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/12/6 0:00:00","PubModel":"Epub","JCR":"Q4","JCRName":"IMMUNOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Introduction: The main pathological feature of osteoarthritis (OA) is chondrocyte injury. LncRNA mitochondrial RNA processing endoribonuclease (RMRP) has been shown to be a chondrogenic differentiation factor. This study aimed to explore the role of RMRP in chondrocyte injury.
Material and methods: Cell counting kit-8 (CCK-8) and TUNEL assays were used to determine lipopolysaccharide (LPS)-induced chondrocyte viability and apoptosis, respectively. The interaction between RMRP and FOXC1 was analyzed by RIP and RNA pull-down. Dual luciferase reporter and ChIP were employed to analyze the interaction between FOXC1 and RBP4. The levels of RMRP, FOXC1, RBP4, apoptosis-related and extracellular matrix (ECM)-related genes were detected by RT-qPCR and western blot. ELISA assay was used for detection of inflammatory cytokines in LPS-induced chondrocytes.
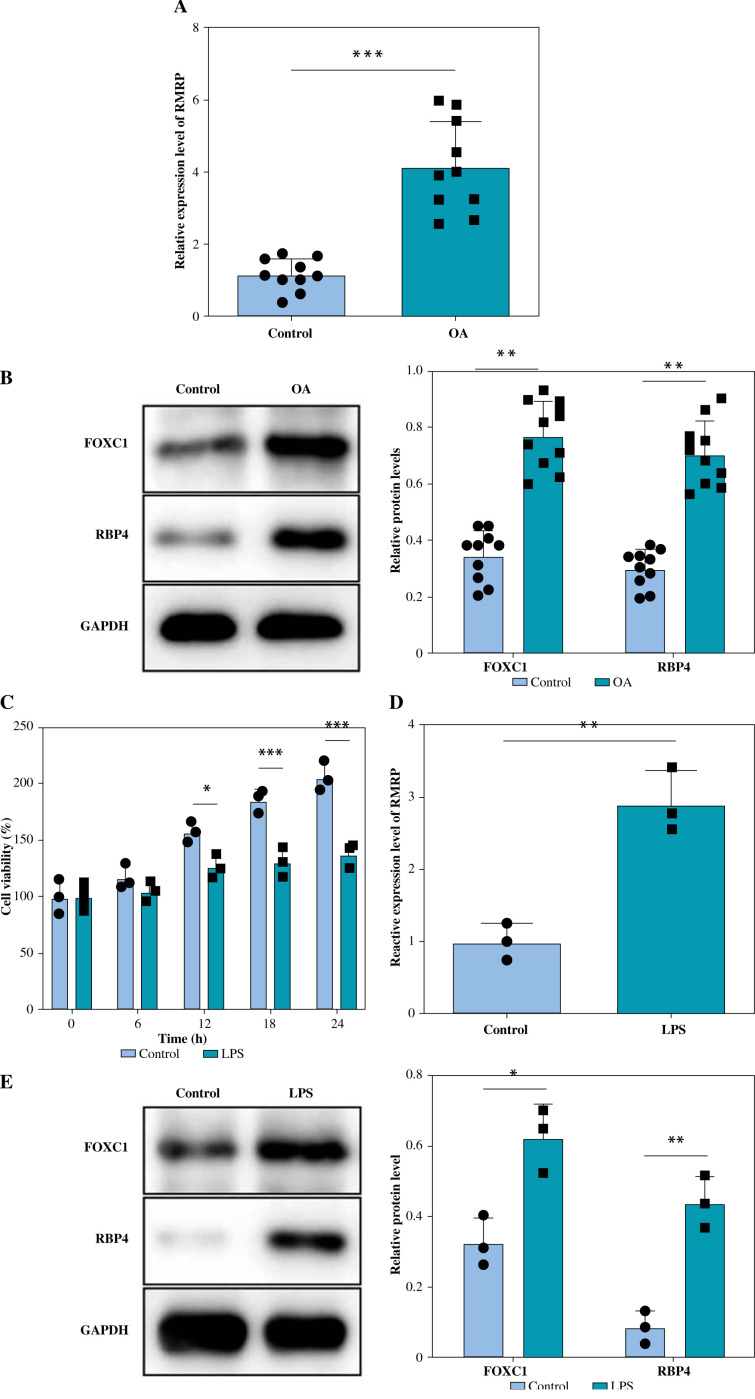
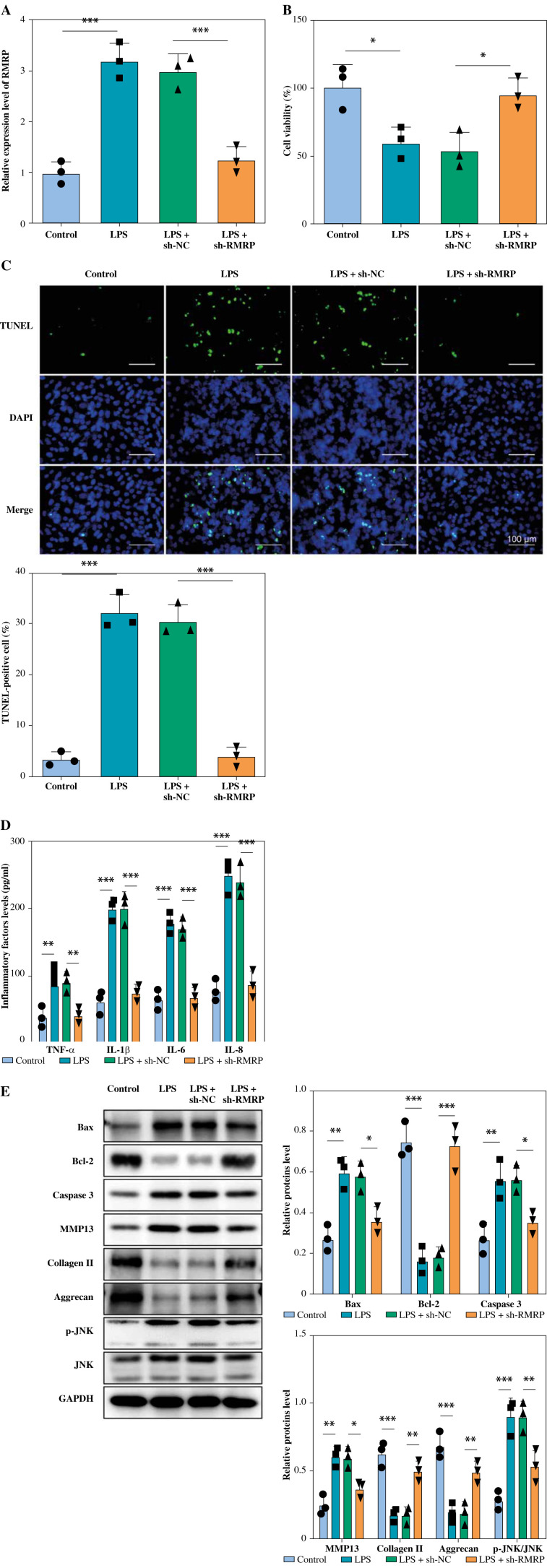
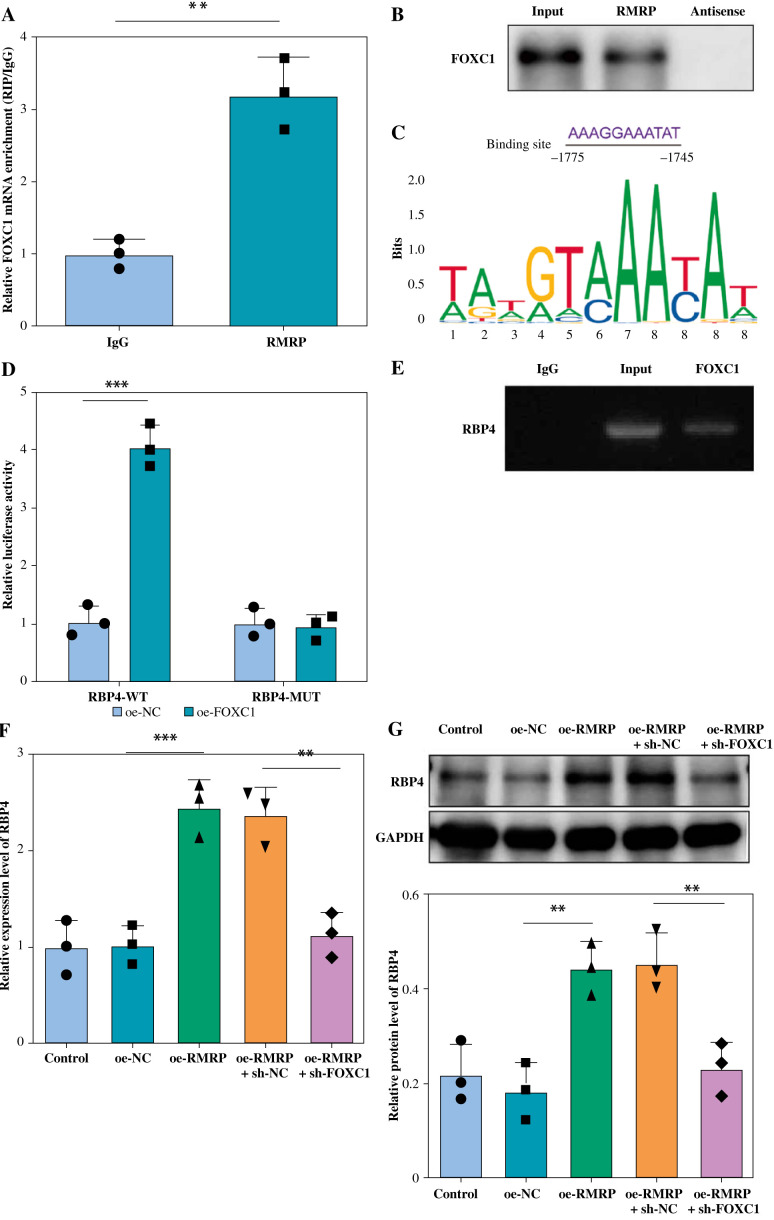
Results: The levels of RMRP, FOXC1 and RBP4 were significantly upregulated in OA cartilage tissues and LPS-induced chondrocytes. Knockdown of RMRP inhibited chondrocyte apoptosis and inflammation under LPS. RMRP interacted with FOXC1 and promoted RBP4 expression. FOXC1 could upregulate RBP4 and promote LPS-induced chondrocyte apoptosis and inflammation. Similarly, RMRP combined with FOXC1 and aggravated apoptosis and inflammation in LPS-treated chondrocytes.
Conclusions: RMRP promoted upregulation of RBP4 and activation of the JNK signaling pathway by binding to FOXC1, thereby accelerating LPS-induced apoptosis and inflammation in chondrocytes.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


