Investigating the effect of active site's coordination number on the oxygen reduction reaction activity of Fe-Co dual-atom catalysts: A theoretical study
IF 8.3
2区 工程技术
Q1 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
Dual-atom catalysts (DACs) have great potential to revolutionize electrocatalytic oxygen reduction reaction (ORR) by utilizing two adjacent active sites to cleave the O-O bond in O2 molecules, but their full potential is limited by insufficient understanding of the coordination environment and electronic interactions between the active sites. Herein, we conducted a comprehensive density functional theory (DFT) analysis on DACs with two coordination configurations, FeN3CoN3 (3N-coordination) and FeN4CoN4 (4N-coordination), revealing how the coordination number around active sites significantly affects ORR performance. The results indicated that the dissociative O2 adsorption configuration, facilitated by cis-bridged O2 adduct formation on Fe and Co active sites of both DACs, was more thermodynamically and kinetically favourable than the associative mechanism on both DACs. The results of free energy calculations indicated that the of FeN3CoN3 is lower than FeN4CoN4, demonstrating that the 3N-coordination environment endowed FeN3CoN3 with a higher adsorption ability as compared to FeN4CoN4 with a 4N-coordination environment. The elimination of ∗OH from the active site, required 0.28 eV and 0.36 eV as the limiting potentials for the FeN4CoN4 and FeN3CoN3 models, respectively, both of which are considerably lower than the 0.68 V (FeN4CoN4) and 0.86 V (FeN3CoN3) observed in single active sites during associative mechanism. Thus, the cis-bridge O2 configuration across neighbouring sites in the DACs significantly reduces the overpotential, demonstrating FeN4CoN4 as a more effective catalytic model than FeN3CoN3. Further, the findings reveal that the 4N-coordination in FeN4CoN4 enhances orbital coupling and spin polarization, boosting its ORR activity compared to FeN3CoN3. The study emphasizes the importance of coordination number in DAC design, highlighting its role in optimizing electrocatalytic performance.
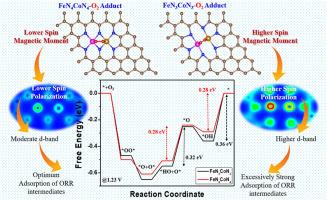
活性位配位数对Fe-Co双原子催化剂氧还原反应活性影响的理论研究
双原子催化剂(DACs)利用两个相邻的活性位点裂解O2分子中的O-O键,在电催化氧还原反应(ORR)方面具有巨大的潜力,但由于对活性位点之间的配位环境和电子相互作用的认识不足,其充分发挥潜力受到限制。本文对具有FeN3CoN3 (3n -配位)和FeN4CoN4 (4n -配位)两种配位构型的dac进行了密度泛函理论(DFT)分析,揭示了活性位点周围的配位数如何显著影响ORR性能。结果表明,在这两种DACs的Fe和Co活性位点上形成顺式桥接O2加合物所促进的解离O2吸附构型,在热力学和动力学上都比两种DACs的缔合机理更有利。自由能计算结果表明,FeN3CoN3的ΔG∗O2低于FeN4CoN4,表明3n配位环境赋予FeN3CoN3比4n配位环境下的FeN4CoN4具有更高的吸附能力。在FeN4CoN4和FeN3CoN3模型中,消除活性位点上的∗OH分别需要0.28 eV和0.36 eV作为极限电位,这两个极限电位都明显低于在结合机制中单个活性位点观察到的0.68 V (FeN4CoN4)和0.86 V (FeN3CoN3)。因此,在dac中相邻位点上的顺式桥式O2结构显著降低了过电位,表明FeN4CoN4是比FeN3CoN3更有效的催化模型。此外,研究结果表明,与FeN3CoN3相比,FeN4CoN4中的4n配位增强了轨道耦合和自旋极化,提高了其ORR活性。本研究强调配位数在DAC设计中的重要性,强调配位数在优化电催化性能中的作用。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

International Journal of Hydrogen Energy
工程技术-环境科学
CiteScore
13.50
自引率
25.00%
发文量
3502
审稿时长
60 days
期刊介绍:
The objective of the International Journal of Hydrogen Energy is to facilitate the exchange of new ideas, technological advancements, and research findings in the field of Hydrogen Energy among scientists and engineers worldwide. This journal showcases original research, both analytical and experimental, covering various aspects of Hydrogen Energy. These include production, storage, transmission, utilization, enabling technologies, environmental impact, economic considerations, and global perspectives on hydrogen and its carriers such as NH3, CH4, alcohols, etc.
The utilization aspect encompasses various methods such as thermochemical (combustion), photochemical, electrochemical (fuel cells), and nuclear conversion of hydrogen, hydrogen isotopes, and hydrogen carriers into thermal, mechanical, and electrical energies. The applications of these energies can be found in transportation (including aerospace), industrial, commercial, and residential sectors.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


