Leveraging Persistent Homology Features for Accurate Defect Formation Energy Predictions via Graph Neural Networks
IF 7
2区 材料科学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
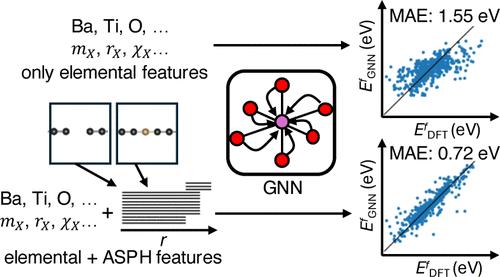
In machine-learning-assisted high-throughput defect studies, a defect-aware latent representation of the supercell structure is crucial for the accurate prediction of defect properties. The performance of current graph neural network (GNN) models is limited due to the fact that defect properties depend strongly on the local atomic configurations near the defect sites and due to the oversmoothing problem of GNN. Herein, we demonstrate that persistent homology features, which encode the topological information on the local chemical environment around each atomic site, can characterize the structural information on defects. Using the dataset containing a wide spectrum of O-based perovskites with all available vacancies as an example, we show that incorporating the persistent homology features, along with proper choices of graph pooling operations, significantly increases the prediction accuracy, with the MAE reduced by 55%. Those features can be easily integrated into the state-of-the-art GNN models, including the graph Transformer network and the equivariant neural network, and universally improve their performance. Besides, our model also overcomes the convergence issue with respect to the supercell size that was present in previous GNN models. Furthermore, using the datasets of defective BaTiO3 with multiple substitutions and multiple vacancies as examples, our GNN model can also predict the defect–defect interactions accurately. These results suggest that persistent homology features can effectively improve the performance of machine learning models and assist the accelerated discovery of functional defects for technological applications.
利用持久的同源特征,通过图神经网络进行准确的缺陷形成能量预测
在机器学习辅助的高通量缺陷研究中,超级单体结构的缺陷感知潜在表示对于准确预测缺陷特性至关重要。由于缺陷特性强烈依赖于缺陷位置附近的局部原子构型以及GNN的过平滑问题,现有的图神经网络(GNN)模型的性能受到限制。在此,我们证明了持久的同源特征,编码了每个原子位点周围局部化学环境的拓扑信息,可以表征缺陷的结构信息。以包含所有可用空缺的广泛的基于钙钛矿的数据集为例,我们表明,结合持久的同源性特征,以及适当的图池化操作选择,显著提高了预测精度,MAE降低了55%。这些特征可以很容易地集成到最先进的GNN模型中,包括图转换器网络和等变神经网络,并普遍提高它们的性能。此外,我们的模型还克服了以往GNN模型中存在的关于超级单体大小的收敛问题。此外,以具有多个取代和多个空位的缺陷BaTiO3数据集为例,我们的GNN模型也可以准确地预测缺陷-缺陷相互作用。这些结果表明,持续的同源特征可以有效地提高机器学习模型的性能,并有助于加速发现技术应用中的功能缺陷。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Chemistry of Materials
工程技术-材料科学:综合
CiteScore
14.10
自引率
5.80%
发文量
929
审稿时长
1.5 months
期刊介绍:
The journal Chemistry of Materials focuses on publishing original research at the intersection of materials science and chemistry. The studies published in the journal involve chemistry as a prominent component and explore topics such as the design, synthesis, characterization, processing, understanding, and application of functional or potentially functional materials. The journal covers various areas of interest, including inorganic and organic solid-state chemistry, nanomaterials, biomaterials, thin films and polymers, and composite/hybrid materials. The journal particularly seeks papers that highlight the creation or development of innovative materials with novel optical, electrical, magnetic, catalytic, or mechanical properties. It is essential that manuscripts on these topics have a primary focus on the chemistry of materials and represent a significant advancement compared to prior research. Before external reviews are sought, submitted manuscripts undergo a review process by a minimum of two editors to ensure their appropriateness for the journal and the presence of sufficient evidence of a significant advance that will be of broad interest to the materials chemistry community.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


