Lamiae Azouggagh, Noelia Ibáñez-Escriche, Marina Martínez-Álvaro, Luis Varona, Joaquim Casellas, Sara Negro, Cristina Casto-Rebollo
{"title":"Characterization of microbiota signatures in Iberian pig strains using machine learning algorithms.","authors":"Lamiae Azouggagh, Noelia Ibáñez-Escriche, Marina Martínez-Álvaro, Luis Varona, Joaquim Casellas, Sara Negro, Cristina Casto-Rebollo","doi":"10.1186/s42523-025-00378-z","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>There is a growing interest in uncovering the factors that shape microbiome composition due to its association with complex phenotypic traits in livestock. Host genetic variation is increasingly recognized as a major factor influencing the microbiome. The Iberian pig breed, known for its high-quality meat products, includes various strains with recognized genetic and phenotypic variability. However, despite the microbiome's known impact on pigs' productive phenotypes such as meat quality traits, comparative analyses of gut microbial composition across Iberian pig strains are lacking. This study aims to explore the gut microbiota of two Iberian pig strains, Entrepelado (n = 74) and Retinto (n = 63), and their reciprocal crosses (n = 100), using machine learning (ML) models to identify key microbial taxa relevant for distinguishing their genetic backgrounds, which holds potential application in the pig industry. Nine ML algorithms, including tree-based, kernel-based, probabilistic, and linear algorithms, were used.</p><p><strong>Results: </strong>Beta diversity analysis on 16 S rRNA microbiome data revealed compositional divergence among genetic, age and batch groups. ML models exploring maternal, paternal and heterosis effects showed varying levels of classification performance, with the paternal effect scenario being the best, achieving a mean Area Under the ROC curve (AUROC) of 0.74 using the Catboost (CB) algorithm. However, the most genetically distant animals, the purebreds, were more easily discriminated using the ML models. The classification of the two Iberian strains reached the highest mean AUROC of 0.83 using Support Vector Machine (SVM) model. The most relevant genera in this classification performance were Acetitomaculum, Butyricicoccus and Limosilactobacillus. All of which exhibited a relevant differential abundance between purebred animals using a Bayesian linear model.</p><p><strong>Conclusions: </strong>The study confirms variations in gut microbiota among Iberian pig strains and their crosses, influenced by genetic and non-genetic factors. ML models, particularly CB and RF, as well as SVM in certain scenarios, combined with a feature selection process, effectively classified genetic groups based on microbiome data and identified key microbial taxa. These taxa were linked to short-chain fatty acids production and lipid metabolism, suggesting microbial composition differences may contribute to variations in fat-related traits among Iberian genetic groups.</p>","PeriodicalId":72201,"journal":{"name":"Animal microbiome","volume":"7 1","pages":"13"},"PeriodicalIF":4.4000,"publicationDate":"2025-02-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11789298/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Animal microbiome","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s42523-025-00378-z","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MICROBIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Background: There is a growing interest in uncovering the factors that shape microbiome composition due to its association with complex phenotypic traits in livestock. Host genetic variation is increasingly recognized as a major factor influencing the microbiome. The Iberian pig breed, known for its high-quality meat products, includes various strains with recognized genetic and phenotypic variability. However, despite the microbiome's known impact on pigs' productive phenotypes such as meat quality traits, comparative analyses of gut microbial composition across Iberian pig strains are lacking. This study aims to explore the gut microbiota of two Iberian pig strains, Entrepelado (n = 74) and Retinto (n = 63), and their reciprocal crosses (n = 100), using machine learning (ML) models to identify key microbial taxa relevant for distinguishing their genetic backgrounds, which holds potential application in the pig industry. Nine ML algorithms, including tree-based, kernel-based, probabilistic, and linear algorithms, were used.
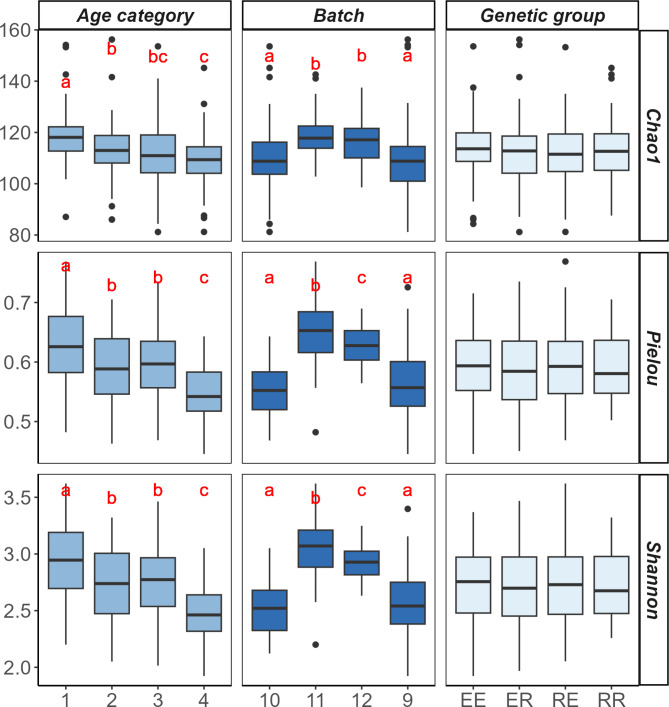
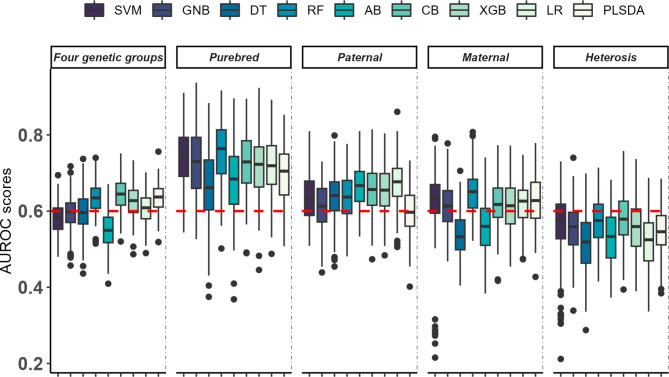
Results: Beta diversity analysis on 16 S rRNA microbiome data revealed compositional divergence among genetic, age and batch groups. ML models exploring maternal, paternal and heterosis effects showed varying levels of classification performance, with the paternal effect scenario being the best, achieving a mean Area Under the ROC curve (AUROC) of 0.74 using the Catboost (CB) algorithm. However, the most genetically distant animals, the purebreds, were more easily discriminated using the ML models. The classification of the two Iberian strains reached the highest mean AUROC of 0.83 using Support Vector Machine (SVM) model. The most relevant genera in this classification performance were Acetitomaculum, Butyricicoccus and Limosilactobacillus. All of which exhibited a relevant differential abundance between purebred animals using a Bayesian linear model.
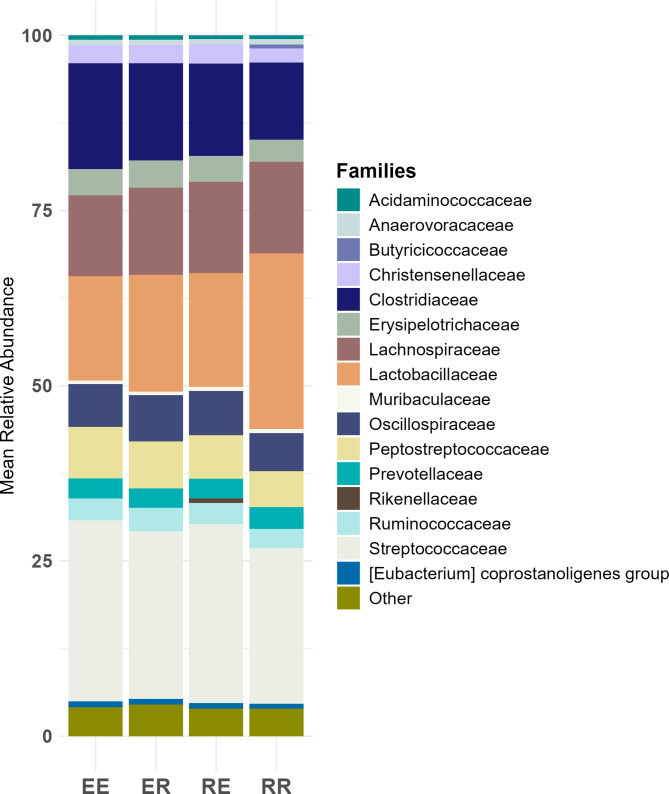
Conclusions: The study confirms variations in gut microbiota among Iberian pig strains and their crosses, influenced by genetic and non-genetic factors. ML models, particularly CB and RF, as well as SVM in certain scenarios, combined with a feature selection process, effectively classified genetic groups based on microbiome data and identified key microbial taxa. These taxa were linked to short-chain fatty acids production and lipid metabolism, suggesting microbial composition differences may contribute to variations in fat-related traits among Iberian genetic groups.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


