Elena Di Simone, Gianvito Vilé, Giovanni Di Liberto* and Gianfranco Pacchioni*,
{"title":"Decoding the Role of Adsorbates Entropy in the Reactivity of Single-Atom Catalysts","authors":"Elena Di Simone, Gianvito Vilé, Giovanni Di Liberto* and Gianfranco Pacchioni*, ","doi":"10.1021/acscatal.4c0447210.1021/acscatal.4c04472","DOIUrl":null,"url":null,"abstract":"<p >Single-atom catalysts (SACs) are rapidly gaining attention as a versatile class of materials that combine the advantages of both homogeneous and heterogeneous catalysis. A growing number of studies aim to identify potential new SACs or to describe their structure and reactivity through ab initio quantum chemical simulations. While many computational studies primarily address reactions involving small molecules, such as water splitting or CO<sub>2</sub> reduction, the application scope of SACs is rapidly broadening to include the production of fine chemicals and the conversion of biomass-derived platform molecules, processes that involve larger, more complex reactants. Using density-functional theory (DFT) simulations, we demonstrate that, while an approximate treatment of entropy is acceptable for molecules with up to three atoms, it introduces substantial errors in reactions involving more complex molecules. Our results reveal a linear correlation between the entropy of adsorbed molecules and that of the corresponding isolated species, mirroring trends observed on extended catalytic surfaces. For the largest systems investigated in this study, the entropy of the free molecule is reduced by approximately 10–20% upon adsorption; for small molecules, this reduction can range from 50 to 70%. This disparity arises because, on SACs, the translational entropy is effectively zero, the rotational entropy is minimal, and the vibrational entropy increases with the size of the molecule. Moreover, the entropy of adsorbates scales linearly with the number of atoms in the molecule, allowing for the prediction of entropic contributions of adsorbates on SACs without additional computational cost. Using propyne hydrogenation as a test, we demonstrate that the reaction energy profile computed with current approximate approaches for estimating the entropy of adsorbates differs significantly from the profile where entropy is explicitly included. These findings highlight the importance of considering adsorbate entropy for accurately predicting the catalytic activity of SACs, particularly for reactions involving complex molecules.</p>","PeriodicalId":9,"journal":{"name":"ACS Catalysis ","volume":"15 1","pages":"447–456 447–456"},"PeriodicalIF":13.1000,"publicationDate":"2024-12-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"ACS Catalysis ","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acscatal.4c04472","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
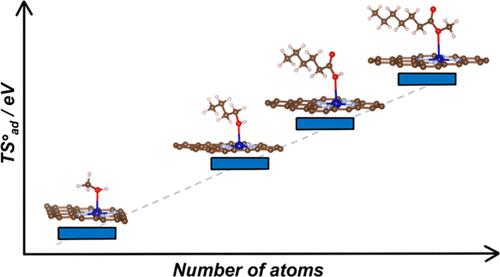
Single-atom catalysts (SACs) are rapidly gaining attention as a versatile class of materials that combine the advantages of both homogeneous and heterogeneous catalysis. A growing number of studies aim to identify potential new SACs or to describe their structure and reactivity through ab initio quantum chemical simulations. While many computational studies primarily address reactions involving small molecules, such as water splitting or CO2 reduction, the application scope of SACs is rapidly broadening to include the production of fine chemicals and the conversion of biomass-derived platform molecules, processes that involve larger, more complex reactants. Using density-functional theory (DFT) simulations, we demonstrate that, while an approximate treatment of entropy is acceptable for molecules with up to three atoms, it introduces substantial errors in reactions involving more complex molecules. Our results reveal a linear correlation between the entropy of adsorbed molecules and that of the corresponding isolated species, mirroring trends observed on extended catalytic surfaces. For the largest systems investigated in this study, the entropy of the free molecule is reduced by approximately 10–20% upon adsorption; for small molecules, this reduction can range from 50 to 70%. This disparity arises because, on SACs, the translational entropy is effectively zero, the rotational entropy is minimal, and the vibrational entropy increases with the size of the molecule. Moreover, the entropy of adsorbates scales linearly with the number of atoms in the molecule, allowing for the prediction of entropic contributions of adsorbates on SACs without additional computational cost. Using propyne hydrogenation as a test, we demonstrate that the reaction energy profile computed with current approximate approaches for estimating the entropy of adsorbates differs significantly from the profile where entropy is explicitly included. These findings highlight the importance of considering adsorbate entropy for accurately predicting the catalytic activity of SACs, particularly for reactions involving complex molecules.
期刊介绍:
ACS Catalysis is an esteemed journal that publishes original research in the fields of heterogeneous catalysis, molecular catalysis, and biocatalysis. It offers broad coverage across diverse areas such as life sciences, organometallics and synthesis, photochemistry and electrochemistry, drug discovery and synthesis, materials science, environmental protection, polymer discovery and synthesis, and energy and fuels.
The scope of the journal is to showcase innovative work in various aspects of catalysis. This includes new reactions and novel synthetic approaches utilizing known catalysts, the discovery or modification of new catalysts, elucidation of catalytic mechanisms through cutting-edge investigations, practical enhancements of existing processes, as well as conceptual advances in the field. Contributions to ACS Catalysis can encompass both experimental and theoretical research focused on catalytic molecules, macromolecules, and materials that exhibit catalytic turnover.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


