H Georg Schulze, Shreyas Rangan, Martha Z Vardaki, Michael W Blades, Robin F B Turner, James M Piret
{"title":"Demixing and Analysis of Complex Biological Raman Hyperspectra Based on Peak Fitting, Amplitude Trend Clustering, and Spectrum Reconstruction.","authors":"H Georg Schulze, Shreyas Rangan, Martha Z Vardaki, Michael W Blades, Robin F B Turner, James M Piret","doi":"10.1177/00037028241311296","DOIUrl":null,"url":null,"abstract":"<p><p>To better interpret the Raman spectra from mammalian cells, it is often desirable to reduce their complexity by decomposing them into the spectral contributions from individual macromolecules or types of macromolecules. Diverse methods exist for demixing complex spectra, each with different benefits and drawbacks. However, some methods require a library of component spectra that might not be available, while others are hampered by noise and peak congestion that includes many proximal overlapping peaks. Through rapid fitting of individual peaks in every spectrum of a Raman hyperspectral data set, we have obtained individual peak parameters from which we determined the trends for all the peak amplitudes. We then grouped similar trends with <i>k</i>-means clustering. Then we used the peak parameters of all the peaks in a given cluster to reconstruct a spectrum representative of that cluster. This method produced spectra that were less distorted by unrelated overlapping peaks or noise, were less congested than those in the hyperspectral set, and thereby improved peak identification and macromolecule recognition. We have demonstrated the application of the method with Raman spectra from a perchlorate-polystyrene model system and extended it to complex spectra from methanol-fixed mammalian cells. We were able to recover independent spectra of perchlorate and polystyrene in the model system and spectra pertaining to individual macromolecular types (proteins, nucleic acids, lipids) from the mammalian cell data. We discuss how imperfections in spectral preprocessing and peak fitting can adversely affect the results. In summary, we have provided a proof-of-concept for a novel mixture resolution method with different attributes than extant ones.</p>","PeriodicalId":8253,"journal":{"name":"Applied Spectroscopy","volume":" ","pages":"1303-1312"},"PeriodicalIF":2.2000,"publicationDate":"2025-09-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12426328/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Applied Spectroscopy","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1177/00037028241311296","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/2/2 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"INSTRUMENTS & INSTRUMENTATION","Score":null,"Total":0}
引用次数: 0
Abstract
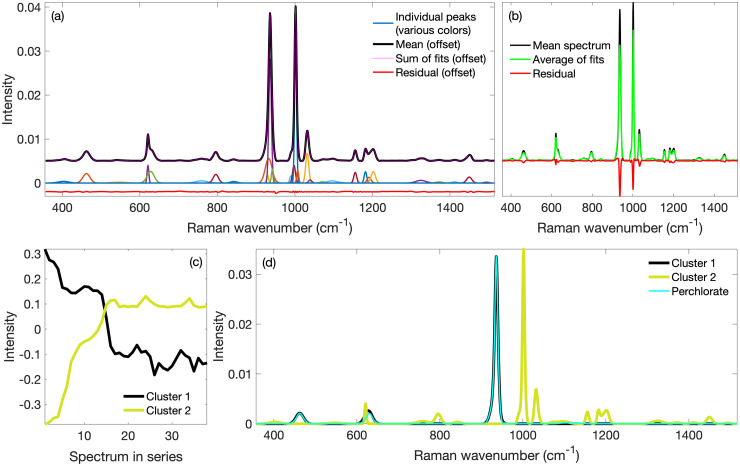
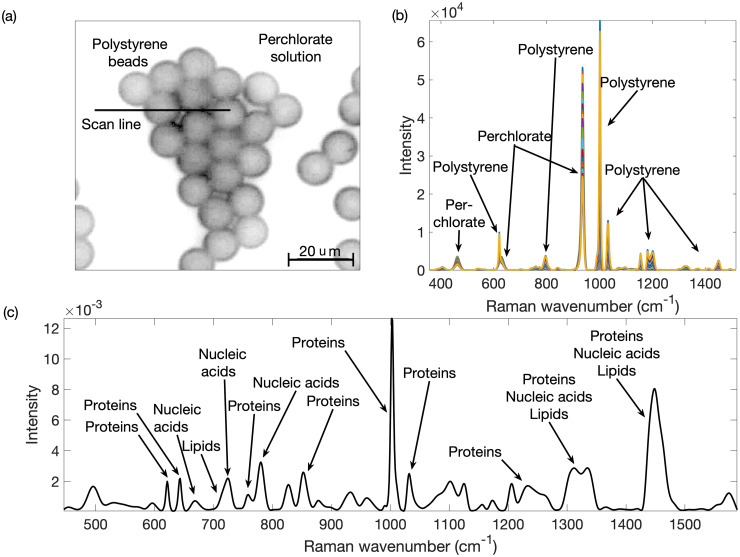
To better interpret the Raman spectra from mammalian cells, it is often desirable to reduce their complexity by decomposing them into the spectral contributions from individual macromolecules or types of macromolecules. Diverse methods exist for demixing complex spectra, each with different benefits and drawbacks. However, some methods require a library of component spectra that might not be available, while others are hampered by noise and peak congestion that includes many proximal overlapping peaks. Through rapid fitting of individual peaks in every spectrum of a Raman hyperspectral data set, we have obtained individual peak parameters from which we determined the trends for all the peak amplitudes. We then grouped similar trends with k-means clustering. Then we used the peak parameters of all the peaks in a given cluster to reconstruct a spectrum representative of that cluster. This method produced spectra that were less distorted by unrelated overlapping peaks or noise, were less congested than those in the hyperspectral set, and thereby improved peak identification and macromolecule recognition. We have demonstrated the application of the method with Raman spectra from a perchlorate-polystyrene model system and extended it to complex spectra from methanol-fixed mammalian cells. We were able to recover independent spectra of perchlorate and polystyrene in the model system and spectra pertaining to individual macromolecular types (proteins, nucleic acids, lipids) from the mammalian cell data. We discuss how imperfections in spectral preprocessing and peak fitting can adversely affect the results. In summary, we have provided a proof-of-concept for a novel mixture resolution method with different attributes than extant ones.
期刊介绍:
Applied Spectroscopy is one of the world''s leading spectroscopy journals, publishing high-quality peer-reviewed articles, both fundamental and applied, covering all aspects of spectroscopy. Established in 1951, the journal is owned by the Society for Applied Spectroscopy and is published monthly. The journal is dedicated to fulfilling the mission of the Society to “…advance and disseminate knowledge and information concerning the art and science of spectroscopy and other allied sciences.”

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


