A barley pan-transcriptome reveals layers of genotype-dependent transcriptional complexity
IF 31.7
1区 生物学
Q1 GENETICS & HEREDITY
引用次数: 0
Abstract
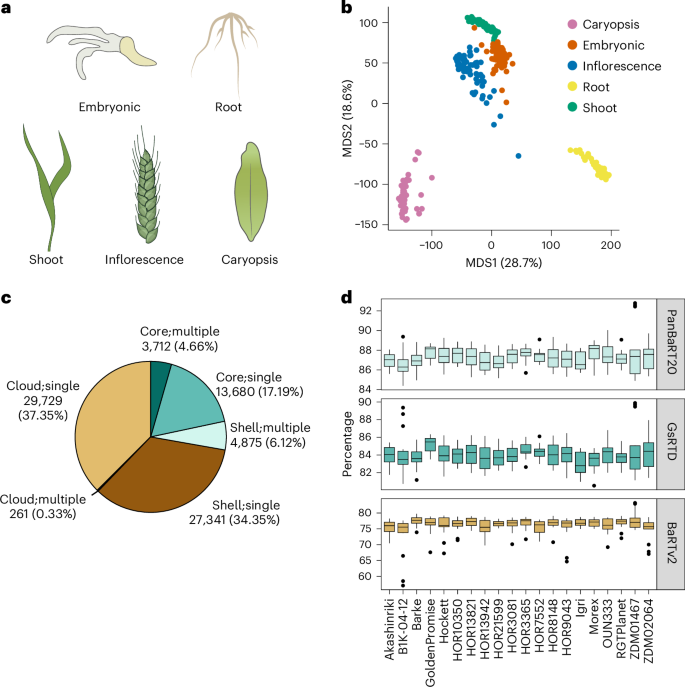
A pan-transcriptome describes the transcriptional and post-transcriptional consequences of genome diversity from multiple individuals within a species. We developed a barley pan-transcriptome using 20 inbred genotypes representing domesticated barley diversity by generating and analyzing short- and long-read RNA-sequencing datasets from multiple tissues. To overcome single reference bias in transcript quantification, we constructed genotype-specific reference transcript datasets (RTDs) and integrated these into a linear pan-genome framework to create a pan-RTD, allowing transcript categorization as core, shell or cloud. Focusing on the core (expressed in all genotypes), we observed significant transcript abundance variation among tissues and between genotypes driven partly by RNA processing, gene copy number, structural rearrangements and conservation of promotor motifs. Network analyses revealed conserved co-expression module::tissue correlations and frequent functional diversification. To complement the pan-transcriptome, we constructed a comprehensive cultivar (cv.) Morex gene-expression atlas and illustrate how these combined datasets can be used to guide biological inquiry. A long- and short-read barley pan-transcriptome assembled from 20 diverse barley genotypes offers insights into genotype- and tissue-dependent gene expression and function.

大麦泛转录组揭示了基因型依赖的转录复杂性层
泛转录组描述了一个物种内多个个体基因组多样性的转录和转录后结果。通过生成和分析来自多个组织的短读和长读rna测序数据集,我们利用代表驯化大麦多样性的20个自交基因型构建了大麦泛转录组。为了克服转录本量化中的单参考偏差,我们构建了基因型特异性参考转录本数据集(rtd),并将其整合到一个线性泛基因组框架中,创建了一个泛rtd,允许转录本分类为核心、壳或云。关注核心(在所有基因型中表达),我们观察到组织之间和基因型之间转录物丰度的显著差异,部分原因是RNA加工、基因拷贝数、结构重排和启动子基序的保护。网络分析揭示了保守的共表达模块:组织相关性和频繁的功能多样化。为了补充泛转录组,我们构建了一个综合品种(cv。Morex基因表达图谱,并说明这些组合数据集如何用于指导生物学探究。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Nature genetics
生物-遗传学
CiteScore
43.00
自引率
2.60%
发文量
241
审稿时长
3 months
期刊介绍:
Nature Genetics publishes the very highest quality research in genetics. It encompasses genetic and functional genomic studies on human and plant traits and on other model organisms. Current emphasis is on the genetic basis for common and complex diseases and on the functional mechanism, architecture and evolution of gene networks, studied by experimental perturbation.
Integrative genetic topics comprise, but are not limited to:
-Genes in the pathology of human disease
-Molecular analysis of simple and complex genetic traits
-Cancer genetics
-Agricultural genomics
-Developmental genetics
-Regulatory variation in gene expression
-Strategies and technologies for extracting function from genomic data
-Pharmacological genomics
-Genome evolution
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


