Abdul Muhsen Abdeen, Jowan Al-Nusair, Malik Samardali, Mohamed Alshal, Amro Al-Astal, Zeid Khitan
{"title":"Complement-Mediated Hemolytic Uremic Syndrome Due to MCP/CD46 Mutation: A Case Report.","authors":"Abdul Muhsen Abdeen, Jowan Al-Nusair, Malik Samardali, Mohamed Alshal, Amro Al-Astal, Zeid Khitan","doi":"10.1177/23247096251316364","DOIUrl":null,"url":null,"abstract":"<p><p>Thrombotic microangiopathy (TMA) is a severe condition characterized by microangiopathic hemolytic anemia, thrombocytopenia, and end-organ damage, often involving the kidneys. Complement-mediated hemolytic uremic syndrome (cHUS), a rare form of TMA, arises from dysregulated alternative complement pathway activation, frequently due to genetic mutations. We report the case of a 23-year-old male presenting with TMA secondary to a heterozygous mutation in the membrane cofactor protein (MCP/CD46) gene. The patient exhibited severe renal and cardiovascular complications, including acute kidney injury requiring hemodialysis, uremic pericarditis, and persistent anemia. Diagnostic evaluation confirmed complement dysregulation, and management with eculizumab, plasmapheresis, and hemodialysis was initiated. Renal biopsy revealed classic TMA features, and genetic testing identified the MCP mutation, underscoring the importance of genetic predispositions in guiding diagnosis and therapy. This case emphasizes the critical role of genetic testing in TMA evaluation and highlights the potential for improved outcomes through targeted complement inhibition and individualized care strategies.</p>","PeriodicalId":16198,"journal":{"name":"Journal of investigative medicine high impact case reports","volume":"13 ","pages":"23247096251316364"},"PeriodicalIF":0.8000,"publicationDate":"2025-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11773514/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of investigative medicine high impact case reports","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1177/23247096251316364","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"MEDICINE, GENERAL & INTERNAL","Score":null,"Total":0}
引用次数: 0
Abstract
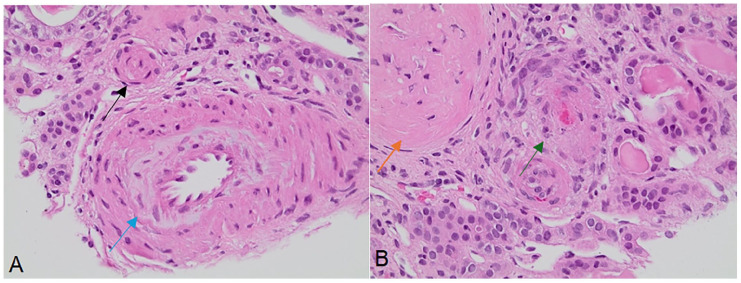
Thrombotic microangiopathy (TMA) is a severe condition characterized by microangiopathic hemolytic anemia, thrombocytopenia, and end-organ damage, often involving the kidneys. Complement-mediated hemolytic uremic syndrome (cHUS), a rare form of TMA, arises from dysregulated alternative complement pathway activation, frequently due to genetic mutations. We report the case of a 23-year-old male presenting with TMA secondary to a heterozygous mutation in the membrane cofactor protein (MCP/CD46) gene. The patient exhibited severe renal and cardiovascular complications, including acute kidney injury requiring hemodialysis, uremic pericarditis, and persistent anemia. Diagnostic evaluation confirmed complement dysregulation, and management with eculizumab, plasmapheresis, and hemodialysis was initiated. Renal biopsy revealed classic TMA features, and genetic testing identified the MCP mutation, underscoring the importance of genetic predispositions in guiding diagnosis and therapy. This case emphasizes the critical role of genetic testing in TMA evaluation and highlights the potential for improved outcomes through targeted complement inhibition and individualized care strategies.
期刊介绍:
The AFMR is committed to enhancing the training and career development of our members and to furthering its mission to facilitate the conduct of research to improve medical care. Case reports represent an important avenue for trainees (interns, residents, and fellows) and early-stage faculty to demonstrate productive, scholarly activity.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


