Jing Lin, Qingwei Ji, Ling Liu, Zongyan Huang, Yingxia Yang, Jie Shen
{"title":"Syncope secondary to arrhythmogenic left ventricular cardiomyopathy: a case report.","authors":"Jing Lin, Qingwei Ji, Ling Liu, Zongyan Huang, Yingxia Yang, Jie Shen","doi":"10.21037/acr-24-131","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Arrhythmogenic cardiomyopathy (ACM) is an inherited cardiac disease characterized by fibrofatty replacement of ventricular myocardium. Ventricular arrhythmia and sudden cardiac death (SCD) are the main clinical manifestations. ACM was previously called arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D). However, recent studies have shown that this disease is not limited to the right ventricle; biventricular involvement occurs in 50% of ACM patients. The left-dominant subtype was subsequently identified, which supported the adoption of the broader term \"ACM\". The clinical literature includes more extensive reports on ARVC, but reports on arrhythmogenic left ventricular cardiomyopathy (ALVC), which is likely to be underrecognized, are limited.</p><p><strong>Case description: </strong>In this report, we describe a case of secondary syncope in a patient with ALVC who developed right bundle branch block with ventricular tachycardia (RBBB-VT), with VT originating in the left ventricle (LV). Cardiac magnetic resonance (CMR) revealed significant enlargement of the LV, with LV dysfunction. Late gadolinium enhancement (LGE) and fat sequencing revealed that most of the free wall of the LV was replaced by fibrofatty tissue.</p><p><strong>Conclusions: </strong>This report could help improve the understanding of this rare disease, and its management. CMR plays a key role in the diagnosis of ACM.</p>","PeriodicalId":29752,"journal":{"name":"AME Case Reports","volume":"9 ","pages":"15"},"PeriodicalIF":0.7000,"publicationDate":"2024-11-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11759933/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"AME Case Reports","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.21037/acr-24-131","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q3","JCRName":"MEDICINE, GENERAL & INTERNAL","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Arrhythmogenic cardiomyopathy (ACM) is an inherited cardiac disease characterized by fibrofatty replacement of ventricular myocardium. Ventricular arrhythmia and sudden cardiac death (SCD) are the main clinical manifestations. ACM was previously called arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D). However, recent studies have shown that this disease is not limited to the right ventricle; biventricular involvement occurs in 50% of ACM patients. The left-dominant subtype was subsequently identified, which supported the adoption of the broader term "ACM". The clinical literature includes more extensive reports on ARVC, but reports on arrhythmogenic left ventricular cardiomyopathy (ALVC), which is likely to be underrecognized, are limited.
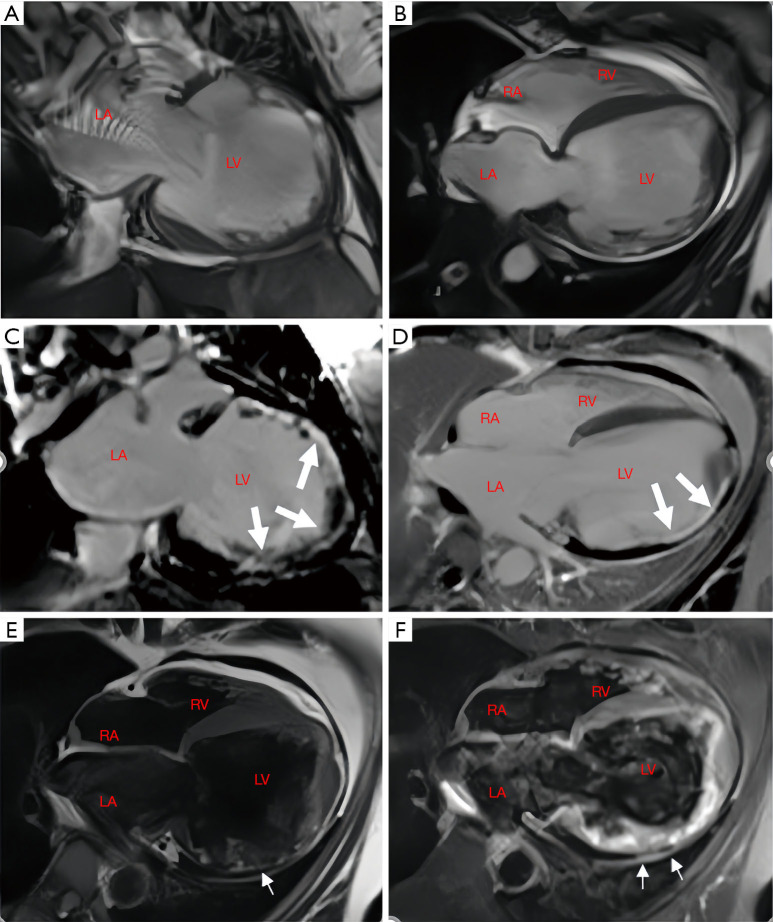
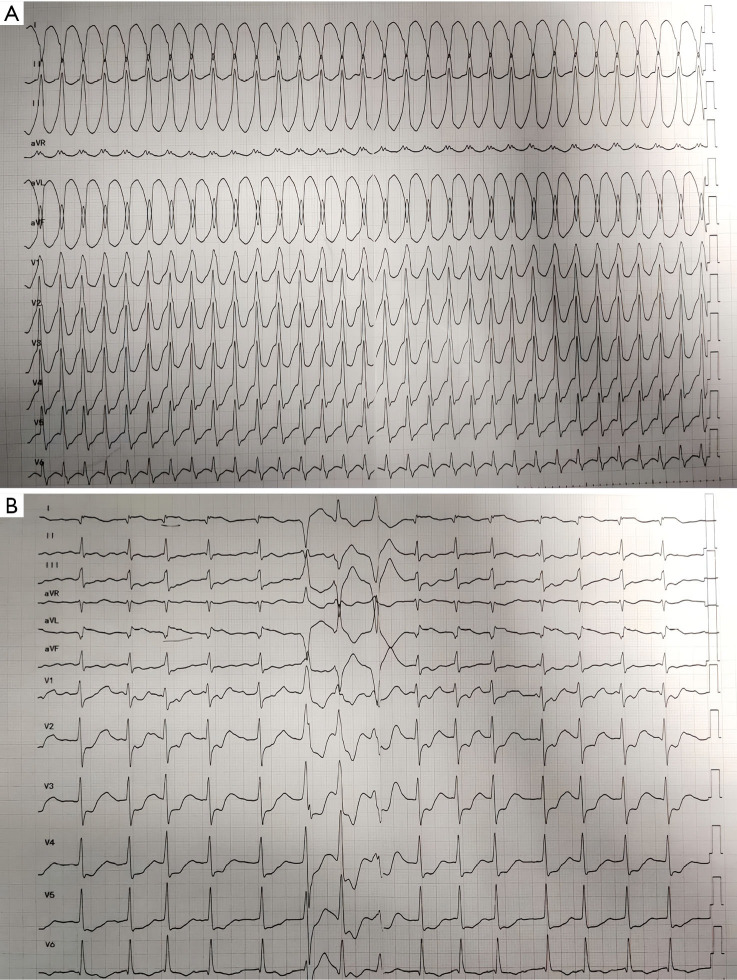
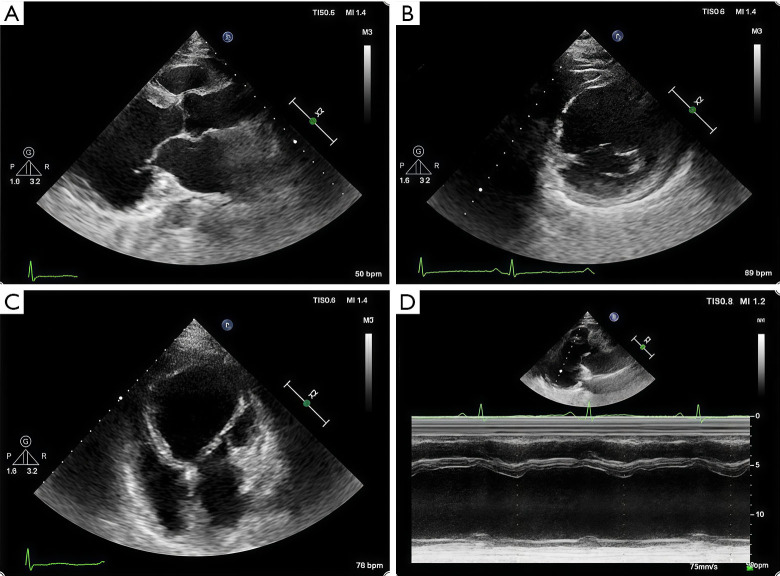
Case description: In this report, we describe a case of secondary syncope in a patient with ALVC who developed right bundle branch block with ventricular tachycardia (RBBB-VT), with VT originating in the left ventricle (LV). Cardiac magnetic resonance (CMR) revealed significant enlargement of the LV, with LV dysfunction. Late gadolinium enhancement (LGE) and fat sequencing revealed that most of the free wall of the LV was replaced by fibrofatty tissue.
Conclusions: This report could help improve the understanding of this rare disease, and its management. CMR plays a key role in the diagnosis of ACM.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


