Adaptive Morphing of Hydroxyl Groups on Covalency Competing Spinel Oxides Boosting Oxygen Evolution Reactions
IF 11.3
1区 化学
Q1 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
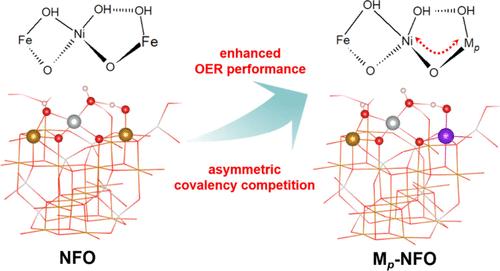
Spinel oxides based on first-row transition metals (Md) have been recognized as cost-effective alternatives to noble metal catalysts in oxygen evolution reactions (OERs), and a multimetal strategy is usually employed to enhance their OER activity. However, it is challenging to rely solely on the combination of these 3d metals to achieve optimal OER activity as their similar metal–oxygen covalency cannot provide sufficient intermodulation to obtain the desirable electronic structure. Here we report that the incorporation of p-block single atoms (Mp, Bi, Sn, and Sb) into octahedral sites of a model spinel oxide NiFe2O4 (NFO) can effectively induce asymmetric Mp-O-Md covalency competition, where the Mp-O covalency acts as an electron reservoir that adaptively modulates the reactivity of Md sites. This allows surface hydroxyl groups to morph dynamically with each electron transfer step, leading to near-optimal formation energies of the OER intermediates. The subsequent experiments as well as in situ and ex situ characterizations have successfully validated the theoretical predictions, especially since the design of Bi-doped NFO (Bi-NFO) shows significant improvement in specific activity compared to pristine spinel NFO. Our work provides important insights into the design principle of earth-abundant oxide catalysts for the OER by leveraging covalency competition, which may be extended to other catalytic reactions.
共价竞争尖晶石氧化物上羟基的自适应变形促进氧进化反应
基于第一排过渡金属(Md)的尖晶石氧化物已被认为是在析氧反应(OERs)中具有成本效益的贵金属催化剂替代品,并且通常采用多金属策略来提高其OER活性。然而,仅仅依靠这些3d金属的组合来实现最佳的OER活性是具有挑战性的,因为它们相似的金属-氧共价不能提供足够的互调来获得理想的电子结构。在这里,我们报道了p-嵌段单原子(Mp, Bi, Sn和Sb)加入到模型尖晶石氧化物NiFe2O4 (NFO)的八面体位点中,可以有效地诱导Mp- o -Md共价竞争,其中Mp- o共价充当电子储集层,自适应调节Md位点的反应性。这使得表面羟基随着每个电子转移步骤动态变形,导致OER中间体的形成能接近最佳。随后的实验以及原位和非原位表征都成功地验证了理论预测,特别是因为与原始尖晶石NFO相比,双掺杂NFO (Bi-NFO)的设计显着提高了比活性。我们的工作通过利用共价竞争为OER的富地氧化物催化剂的设计原则提供了重要的见解,这可能扩展到其他催化反应。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

ACS Catalysis
CHEMISTRY, PHYSICAL-
CiteScore
20.80
自引率
6.20%
发文量
1253
审稿时长
1.5 months
期刊介绍:
ACS Catalysis is an esteemed journal that publishes original research in the fields of heterogeneous catalysis, molecular catalysis, and biocatalysis. It offers broad coverage across diverse areas such as life sciences, organometallics and synthesis, photochemistry and electrochemistry, drug discovery and synthesis, materials science, environmental protection, polymer discovery and synthesis, and energy and fuels.
The scope of the journal is to showcase innovative work in various aspects of catalysis. This includes new reactions and novel synthetic approaches utilizing known catalysts, the discovery or modification of new catalysts, elucidation of catalytic mechanisms through cutting-edge investigations, practical enhancements of existing processes, as well as conceptual advances in the field. Contributions to ACS Catalysis can encompass both experimental and theoretical research focused on catalytic molecules, macromolecules, and materials that exhibit catalytic turnover.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


