Clinical practice recommendations for the diagnosis and management of X-linked hypophosphataemia
IF 39.8
1区 医学
Q1 UROLOGY & NEPHROLOGY
引用次数: 0
Abstract
X-linked hypophosphataemia (XLH) is a rare metabolic bone disorder caused by pathogenic variants in the PHEX gene, which is predominantly expressed in osteoblasts, osteocytes and odontoblasts. XLH is characterized by increased synthesis of the bone-derived phosphaturic hormone fibroblast growth factor 23 (FGF23), which results in renal phosphate wasting with consecutive hypophosphataemia, rickets, osteomalacia, disproportionate short stature, oral manifestations, pseudofractures, craniosynostosis, enthesopathies and osteoarthritis. Patients with XLH should be provided with multidisciplinary care organized by a metabolic bone expert. Historically, these patients were treated with frequent doses of oral phosphate supplements and active vitamin D, which was of limited efficiency and associated with adverse effects. However, the management of XLH has evolved in the past few years owing to the availability of burosumab, a fully humanized monoclonal antibody that neutralizes circulating FGF23. Here, we provide updated clinical practice recommendations for the diagnosis and management of XLH to improve outcomes and quality of life in these patients. The management of X-linked hypophosphataemia has evolved owing to the availability of burosumab. In this Evidence-Based Guideline, the authors provide updated clinical practice recommendations for the diagnosis and management of X-linked hypophosphataemia with the aim of improving patient outcomes and quality of life.

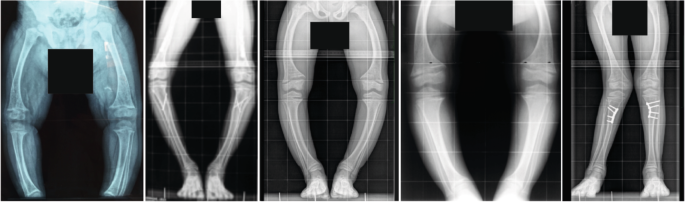
诊断和治疗 X 连锁低磷血症的临床实践建议
x连锁低磷血症(XLH)是一种罕见的代谢性骨疾病,由PHEX基因的致病性变异引起,主要在成骨细胞、骨细胞和成牙细胞中表达。XLH的特点是骨源性磷激素成纤维细胞生长因子23 (FGF23)合成增加,导致肾磷消耗,伴有连续的低磷血症、佝偻病、骨软化症、不成比例的矮小、口腔症状、假性骨折、颅缝闭闭、骨髓瘤病和骨关节炎。XLH患者应接受代谢骨专家组织的多学科治疗。从历史上看,这些患者经常服用口服磷酸盐补充剂和活性维生素D,但效果有限,并伴有不良反应。然而,由于burrosumab(一种完全人源化的单克隆抗体,可中和循环中的FGF23)的可用性,XLH的管理在过去几年中发生了变化。在这里,我们为XLH的诊断和管理提供最新的临床实践建议,以改善这些患者的预后和生活质量。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Nature Reviews Nephrology
医学-泌尿学与肾脏学
CiteScore
39.00
自引率
1.20%
发文量
127
审稿时长
6-12 weeks
期刊介绍:
Nature Reviews Nephrology aims to be the premier source of reviews and commentaries for the scientific communities it serves.
It strives to publish authoritative, accessible articles.
Articles are enhanced with clearly understandable figures, tables, and other display items.
Nature Reviews Nephrology publishes Research Highlights, News & Views, Comments, Reviews, Perspectives, and Consensus Statements.
The content is relevant to nephrologists and basic science researchers.
The broad scope of the journal ensures that the work reaches the widest possible audience.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


