A Density Functional Theory (DFT) Modeling Study of NO Reduction by CO over Graphene-Supported Single-Atom Ni Catalysts in the Presence of CO2, SO2, O2, and H2O
IF 3.9
2区 化学
Q2 CHEMISTRY, MULTIDISCIPLINARY
引用次数: 0
Abstract
The mechanisms of NO reduction by CO over nitrogen-doped graphene (N-graphene)-supported single-atom Ni catalysts in the presence of O2, H2O, CO2, and SO2 have been studied via density functional theory (DFT) modeling. The catalyst is represented by a single Ni atom bonded to four N atoms on N-graphene. Several alternative reaction pathways, including adsorption of NO on the Ni site, direct reduction of NO by CO, decomposition of NO to N2O followed by reduction of N2O to N2, formation of active oxygen radical O*, and reduction of O* by CO, were hypothesized and the energy barrier corresponding to each of the reaction steps was calculated using DFT. The most probable pathway was found to be that NO adsorbed on the Ni site decomposes via the Langmuir–Hinshelwood mechanism to form N2O and subsequently N2, leaving an active oxygen radical (O*) on the surface, which is then reduced by CO. The large adsorption energy of NO on the Ni site results in strong resistance to CO2, SO2, O2, and water vapor. The activation energy of N2O reduction to N2 was found to be larger than those of NO decomposition to N2O and active oxygen radical reduction by CO, illustrating that the step of N2O reduced to N2 is the rate-controlling step.
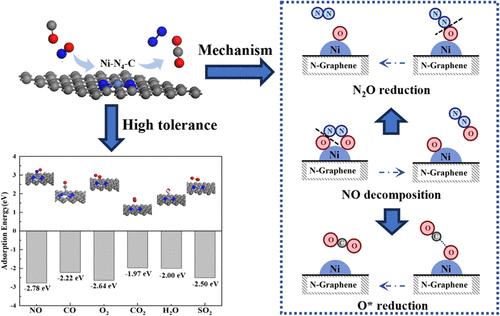
CO2, SO2, O2和H2O存在下,石墨烯负载的单原子Ni催化剂上CO还原NO的密度泛函理论(DFT)建模研究
通过密度泛函理论(DFT)建模研究了掺氮石墨烯(N-石墨烯)支撑的单原子镍催化剂在 O2、H2O、CO2 和 SO2 存在下 CO 还原 NO 的机理。催化剂由一个镍原子与 N-石墨烯上的四个 N 原子键合而成。假设了几种可供选择的反应途径,包括 NO 在 Ni 位点上的吸附、NO 被 CO 直接还原、NO 分解为 N2O,然后 N2O 还原为 N2、活性氧自由基 O* 的形成以及 O* 被 CO 还原。研究发现,最可能的途径是吸附在镍位点上的 NO 通过 Langmuir-Hinshelwood 机理分解为 N2O,随后生成 N2,在表面留下活性氧自由基(O*),然后被 CO 还原。由于 NO 在镍位点上的吸附能很大,因此对 CO2、SO2、O2 和水蒸气具有很强的抗性。研究发现,N2O 还原成 N2 的活化能大于 NO 分解成 N2O 和活性氧自由基被 CO 还原的活化能,这说明 N2O 还原成 N2 的步骤是速率控制步骤。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Langmuir
化学-材料科学:综合
CiteScore
6.50
自引率
10.30%
发文量
1464
审稿时长
2.1 months
期刊介绍:
Langmuir is an interdisciplinary journal publishing articles in the following subject categories:
Colloids: surfactants and self-assembly, dispersions, emulsions, foams
Interfaces: adsorption, reactions, films, forces
Biological Interfaces: biocolloids, biomolecular and biomimetic materials
Materials: nano- and mesostructured materials, polymers, gels, liquid crystals
Electrochemistry: interfacial charge transfer, charge transport, electrocatalysis, electrokinetic phenomena, bioelectrochemistry
Devices and Applications: sensors, fluidics, patterning, catalysis, photonic crystals
However, when high-impact, original work is submitted that does not fit within the above categories, decisions to accept or decline such papers will be based on one criteria: What Would Irving Do?
Langmuir ranks #2 in citations out of 136 journals in the category of Physical Chemistry with 113,157 total citations. The journal received an Impact Factor of 4.384*.
This journal is also indexed in the categories of Materials Science (ranked #1) and Multidisciplinary Chemistry (ranked #5).
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


