Hannah Demond, Soumen Khan, Juan Castillo-Fernandez, Courtney W Hanna, Gavin Kelsey
{"title":"Transcriptome and DNA methylation profiling during the NSN to SN transition in mouse oocytes.","authors":"Hannah Demond, Soumen Khan, Juan Castillo-Fernandez, Courtney W Hanna, Gavin Kelsey","doi":"10.1186/s12860-024-00527-3","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>During the latter stages of their development, mammalian oocytes under dramatic chromatin reconfiguration, transitioning from a non-surrounded nucleolus (NSN) to a surrounded nucleolus (SN) stage, and concomitant transcriptional silencing. Although the NSN-SN transition is known to be essential for developmental competence of the oocyte, less is known about the accompanying molecular changes. Here we examine the changes in the transcriptome and DNA methylation during the NSN to SN transition in mouse oocytes.</p><p><strong>Results: </strong>To study the transcriptome and DNA methylation dynamics during the NSN to SN transition, we used single-cell (sc)M&T-seq to generate scRNA-seq and sc-bisulphite-seq (scBS-seq) data from GV oocytes classified as NSN or SN by Hoechst staining of their nuclei. Transcriptome analysis showed a lower number of detected transcripts in SN compared with NSN oocytes as well as downregulation of 576 genes, which were enriched for processes related to mRNA processing. We used the transcriptome data to generate a classifier that can infer chromatin stage in scRNA-seq datasets. The classifier was successfully tested in multiple published datasets of mouse models with a known skew in NSN: SN ratios. Analysis of the scBS-seq data showed increased DNA methylation in SN compared to NSN oocytes, which was most pronounced in regions with intermediate levels of methylation. Overlap with chromatin immunoprecipitation and sequencing (ChIP-seq) data for the histone modifications H3K36me3, H3K4me3 and H3K27me3 showed that regions gaining methylation in SN oocytes are enriched for overlapping H3K36me3 and H3K27me3, which is an unusual combination, as these marks do not typically coincide.</p><p><strong>Conclusions: </strong>We characterise the transcriptome and DNA methylation changes accompanying the NSN-SN transition in mouse oocytes. We develop a classifier that can be used to infer chromatin status in single-cell or bulk RNA-seq data, enabling identification of altered chromatin transition in genetic knock-outs, and a quality control to identify skewed NSN-SN proportions that could otherwise confound differential gene expression analysis. We identify late-methylating regions in SN oocytes that are associated with an unusual combination of chromatin modifications, which may be regions with high chromatin plasticity and transitioning between H3K27me3 and H3K36me3, or reflect heterogeneity on a single-cell level.</p>","PeriodicalId":9099,"journal":{"name":"BMC Molecular and Cell Biology","volume":"26 1","pages":"2"},"PeriodicalIF":2.7000,"publicationDate":"2025-01-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11697814/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Molecular and Cell Biology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s12860-024-00527-3","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"CELL BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Background: During the latter stages of their development, mammalian oocytes under dramatic chromatin reconfiguration, transitioning from a non-surrounded nucleolus (NSN) to a surrounded nucleolus (SN) stage, and concomitant transcriptional silencing. Although the NSN-SN transition is known to be essential for developmental competence of the oocyte, less is known about the accompanying molecular changes. Here we examine the changes in the transcriptome and DNA methylation during the NSN to SN transition in mouse oocytes.
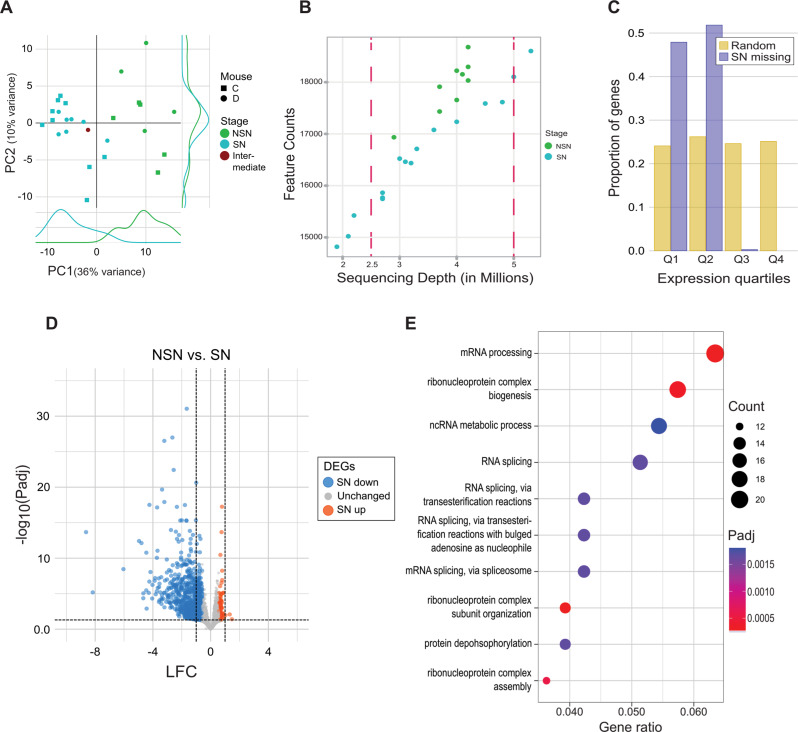
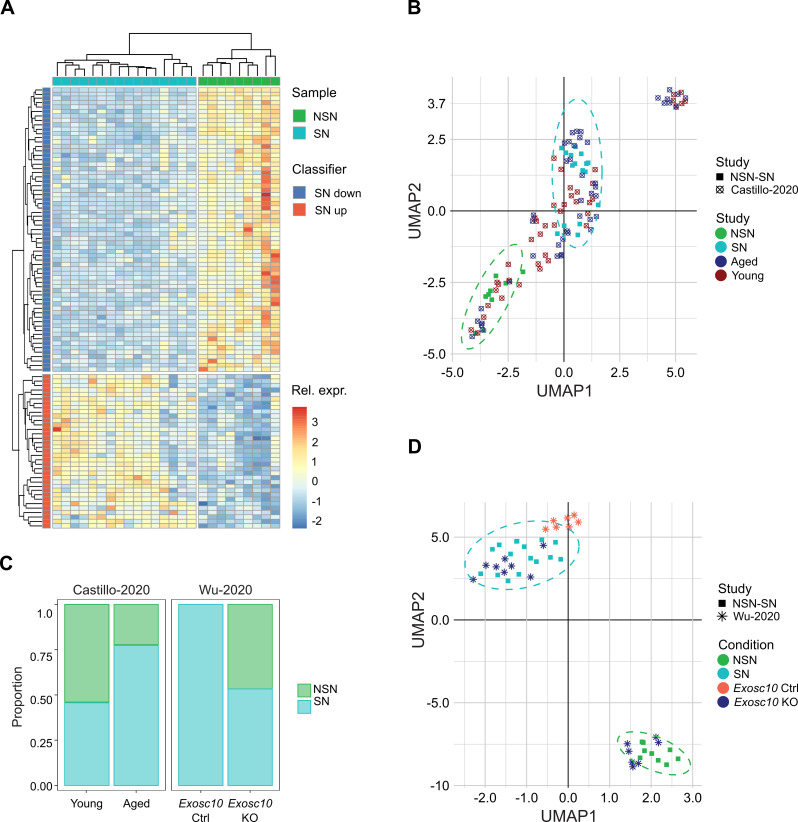
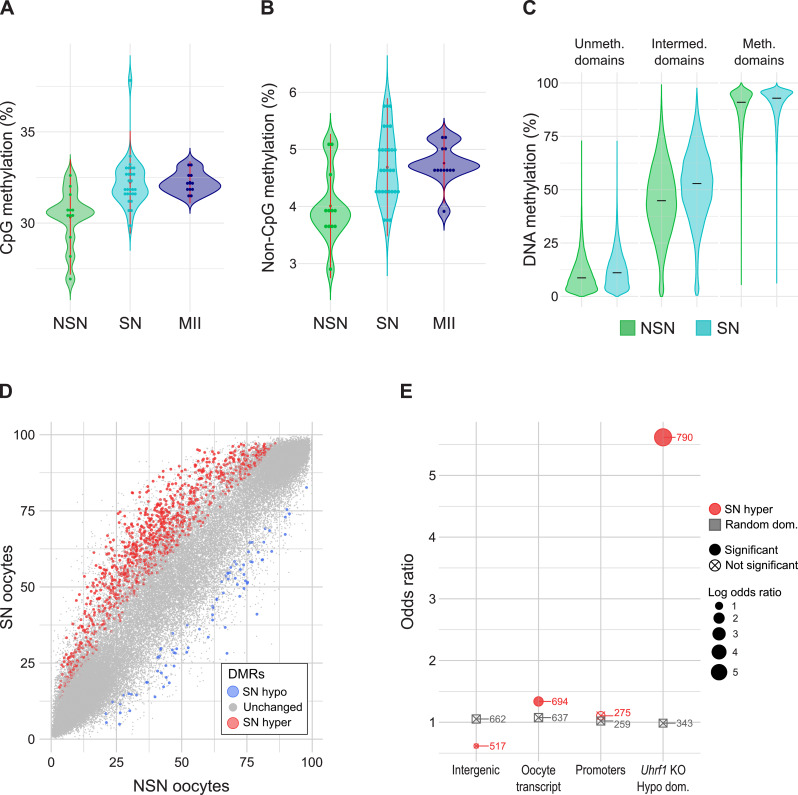
Results: To study the transcriptome and DNA methylation dynamics during the NSN to SN transition, we used single-cell (sc)M&T-seq to generate scRNA-seq and sc-bisulphite-seq (scBS-seq) data from GV oocytes classified as NSN or SN by Hoechst staining of their nuclei. Transcriptome analysis showed a lower number of detected transcripts in SN compared with NSN oocytes as well as downregulation of 576 genes, which were enriched for processes related to mRNA processing. We used the transcriptome data to generate a classifier that can infer chromatin stage in scRNA-seq datasets. The classifier was successfully tested in multiple published datasets of mouse models with a known skew in NSN: SN ratios. Analysis of the scBS-seq data showed increased DNA methylation in SN compared to NSN oocytes, which was most pronounced in regions with intermediate levels of methylation. Overlap with chromatin immunoprecipitation and sequencing (ChIP-seq) data for the histone modifications H3K36me3, H3K4me3 and H3K27me3 showed that regions gaining methylation in SN oocytes are enriched for overlapping H3K36me3 and H3K27me3, which is an unusual combination, as these marks do not typically coincide.
Conclusions: We characterise the transcriptome and DNA methylation changes accompanying the NSN-SN transition in mouse oocytes. We develop a classifier that can be used to infer chromatin status in single-cell or bulk RNA-seq data, enabling identification of altered chromatin transition in genetic knock-outs, and a quality control to identify skewed NSN-SN proportions that could otherwise confound differential gene expression analysis. We identify late-methylating regions in SN oocytes that are associated with an unusual combination of chromatin modifications, which may be regions with high chromatin plasticity and transitioning between H3K27me3 and H3K36me3, or reflect heterogeneity on a single-cell level.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


