Small-Sample Learning for Next-Generation Human Health Risk Assessment: Harnessing AI, Exposome Data, and Systems Biology
IF 10.8
1区 环境科学与生态学
Q1 ENGINEERING, ENVIRONMENTAL
引用次数: 0
Abstract
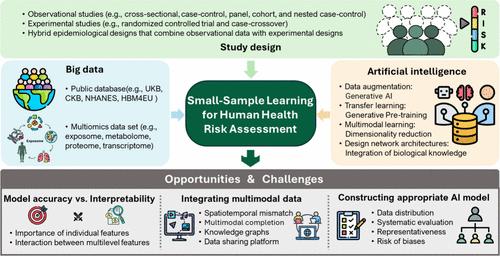
Figure 1. Schematic diagram of small-sample learning for human health risk assessment (HRA) of environmental exposure. To facilitate the related HRA modeling, three aspects should be considered: study design, big data collection, and artificial intelligence. The development of future models should prioritize balancing accuracy with interpretability, integrating multimodal data effectively, and constructing robust, generalizable AI models that incorporate complexity and prior biological knowledge while addressing biases in the data. Utilizing large-scale and reliable human biomonitoring databases can provide prior knowledge on the association between environmental exposure and health outcome. Over the past few decades, several large-scale human biomonitoring studies have been established across various countries and regions to conduct epidemiological research on lifestyle, environmental, and genetic factors that contribute to major diseases. Many of these studies are committed to open data and resource sharing, providing data in a fair and transparent manner. These include projects such as the UK Biobank (UKB), Human Biomonitoring for Europe (HBM4 EU), National Health and Nutrition Examination Survey (NHANES), Human Health Exposure Analysis Resource (HHEAR), China Health and Retirement Longitudinal Study (CHARLS), the Environmental Influences on Child Health Outcomes (ECHO), etc. These databases provide comprehensive data on biospecimens, genetics, imaging, medical health records, lifestyle, air pollution, and chemical exposures, offering invaluable resources for early career researchers to explore and test hypotheses regarding the impact of environmental exposures on human diseases. For example, Argentieri and colleagues developed a biological age prediction model using proteomics data from the UKB (N = 45 441) by identifying 204 key plasma proteins from 2897 proteins for constructing a proteomic age clock. (5) Validation in independent cohorts from China (N = 3977) and Finland (N = 1990) based on the same 204 proteins demonstrated high predictive accuracy. By leveraging biomarkers with strong generalizability identified in large populations, this approach facilitates accurate risk evaluations in smaller cohorts, enhances the identification of vulnerable groups, and helps minimize errors while improving the interpretability of results in small populations. In cases of scarce human biomonitoring data, in vitro to in vivo extrapolation (IVIVE) also enables precise health risk predictions at the individual level for small populations. It also provides confidence for the routine use of chemical prioritization, hazard assessment, and regulatory decision making. Recently, Han et al. systematically summarized and discussed IVIVE methods in next-generation HRA and innovatively proposed prospects from two aspects. (6) The first is to expand the scope of IVIVE, such as focusing on the joint risk of parent compounds and their metabolites; the second is to integrate new technologies like systems biology, multiomics, and adverse outcome networks in IVIVE, aiming at a more microscopic, mechanistic, and comprehensive risk assessment. Incorporating multilevel omics data into small-sample HRA can provide a more comprehensive and accurate understanding of how environmental exposures affect health. It has been well-known that environmental exposures typically influence biological processes at various levels, leading to adverse health outcomes. However, a limitation of multiomics studies is the high cost, with the expense of single-cell transcriptomics ranging from $1500 to $2000 per person, resulting in a typically small-sample size for high-throughput omics assessments. The “Curse of Dimensionality” is a significant modeling challenge in multiomics data. As the dimensionality of the data increases, the sparsity of the data space grows exponentially, causing the occurrence probability of certain feature combinations to become extremely low or even completely unobserved. This sparsity makes it difficult for models to learn meaningful patterns, thereby affecting their generalization ability. Compounding this challenge, multiomics data obtained through different measurement techniques often exhibit distinct characteristics and distributions. (7) Faced with the challenges of small-sample size and high dimensionality, deep learning algorithms can capture complex nonlinear relationships and automatically learn high-quality feature representations from low-level omics data while performing dimensionality reduction. For example, Cao et al. developed a modular framework called GLUE that utilizes a knowledge-based graph to simulate cross-layer regulatory interactions, linking the feature spaces between omics layers. In this graph, the vertices correspond to features from different omics layers and the edges represent regulatory interactions between them. (8) This model was employed to integrate unpaired single-cell multiomics data while simultaneously inferring regulatory interactions, demonstrating exceptional robustness in small-sample scenarios. To mitigate the multicollinearity issue in multiomics data, it is essential to ensure high data quality and diversity, select appropriate model training and regularization strategies, and integrate existing biological knowledge (e.g., gene regulatory networks and protein–protein interactions) to improve model efficiency. Data augmentation. This is a technique used to produce new artificial data that closely resemble real data, thereby enhancing the dataset. In this context, AI-generated content (AIGC) leverages large amounts of unlabeled data to learn data distributions and generate samples, facilitating the creation of images, text, audio, and video while enhancing model generalization capabilities under limited labeled data conditions. Common models include generative adversarial networks (GANs), which generate realistic samples through adversarial training between a generator and a discriminator, leveraging the flexibility of latent space to effectively capture data features. Diffusion models generate high-quality data from random noise through a gradual denoising process, allowing for effective utilization of limited data and producing diverse outputs. Variational autoencoders (VAEs) compress data into latent space via an encoder and then decode it to generate new samples, providing a stable generation process that preserves data diversity and structural integrity in small-sample learning scenarios. AIGCs have made significant advances in molecular property prediction (9) and drug design (10) but remain underexplored in datasets related to HRA. Transfer learning. Machine learning or deep learning models can be trained on large general datasets and fine-tuned with smaller, domain-specific datasets for downstream tasks. One of the key applications of transfer learning in HAR research is using insights from the structure–property relationships of chemicals and shared biological networks underlying diseases to apply knowledge from conventional pollutants to emerging pollutants and from common diseases to rare diseases. Upon application of transfer learning, it is essential to align key factors between the source and target populations, including exposure characteristics (e.g., dose–response relationships), population structures, disease incidence, and disease types. Fine-tuning is crucial in transfer learning and involves strategies like adjusting layer-specific learning rates, freezing early layers while tuning later ones, and using data augmentation or regularization to prevent overfitting. Generative pretraining (GPT), leveraging attention mechanisms and unsupervised pretraining, has become a pioneering model for natural language processing tasks. Inspired by GPT, researchers have applied self-supervised pretraining to large-scale single-cell transcriptomics data to develop foundational models that can be applied to smaller patient data sets, aiding in the identification of candidate therapeutic targets for diseases. (11) In the near future, models similar to ChatGPT may be trained on human biomonitoring databases, although further research is needed to validate these applications. Multimodal learning. In HRA research, the exposure data modalities primarily include numerical vectors (e.g., multiomics data and pollutant concentrations), graphs (e.g., molecular graphs and biological knowledge graphs), text (e.g., electronic health records and protein sequences), visual data (e.g., imaging and remote sensing), and audio data (e.g., environmental noise). In tasks involving systems biology, multimodal learning integrates complementary information from various modalities, enhancing performance under small-sample learning and offering a more comprehensive perspective on environmental health issues. (12) Multimodal learning is a versatile concept that can be addressed with various architectures. A common strategy is to perform joint modeling of data from different modalities, either by designing specialized architectures for each modality or by developing a foundational model that maps similar concepts across modalities into a shared latent space. This approach enables the generation of unified internal representations for the same semantics or concepts (e.g., the unification of remote sensing images, sensor exposure data, and exposure event descriptions) and outputs task-specific results as required. Foundation models have been successfully implemented in integrating single-cell omics data (8) and biomedical data. (13) Another approach is to construct knowledge graphs (KGs) between modalities. In systems biology, multilevel features can be represented as a heterogeneous graph, capturing associations among chemicals, genes, proteins, and diseases, with graph neural networks used to uncover unknown relationships among these entities. Multimodal models also offer a new approach for exposome, integrating external environmental exposure factors with internal exposure biomarkers to create unified feature vectors, which provide a comprehensive individual exposure profile. Sparsely structured network architectures or algorithms. These are designed on the basis of biological information and can reduce model parameters. (14) Biological information design refers to hidden nodes in neural networks that simulate specific biological processes, such as DCell (cellular processes and functions) (15) and P-NET (genes and pathways). (14) More streamlined architectures and sparse network structures enable higher accuracy in small-sample learning, while quantifying the relative importance of biological processes within the network also enhances interpretability. The deep forest model (DF), a multilayer tree-based forest cascade structure, is suitable for data sets of different sizes, few hyperparameters, and adaptive generation of model complexity. The model complexity of DF can be adaptively determined under sufficient training, making it applicable to data sets of small-size scales. (16) Inspired by the adverse outcome pathway (AOP) framework, we can integrate environmental exposures, molecular initiating events (MIEs), key molecular events (KEs), and adverse outcomes into the network architecture, ordered by increasing complexity. (17) It is worth noting that in specific environmental small-sample studies, these methods are not conducted in isolation but often employ a combination of strategies systematically. For example, Huang et al. integrated multimodal data, including genomic data, cellular signaling, gene expression levels, and clinical records, to construct a biomedical knowledge graph and performed self-supervised pretraining. (18) By combining the concepts of multimodal learning and transfer learning, their model was able to accurately predict drug indications and contraindications across diseases under strict zero-shot conditions, including diseases for which no therapeutic drugs are currently available. Balancing model accuracy and interpretability. While deep learning has achieved remarkable success across various fields, interpretability remains a recurrent issue. In the context of HRA, it is especially important to translate model weight parameters into meaningful biological information. This not only provides reliable information to healthcare providers but also offers potential biological explanations for disease progression. Common approaches include SHAP (Shapley additive explanations) and attention weight in transformer architectures. However, in small-sample scenarios, data-driven interpretability mining can sometimes produce spurious important features, leading to interpretations that deviate from biological facts from the in-depth environmental toxicological studies. (19) Another approach is to evaluate key environmental exposure and biological features relevant to disease through computational perturbation of features (e.g., deletion, random masking, or omics data inversion) and quantifying the impact of these perturbations on predictive outcomes. To enhance interpretability, perturbing known features (e.g., MIE and KE) instead of random features can be useful, though this requires prior knowledge related to toxicity pathways. While the approach described above applies to assessing the importance of individual features, the advantage of deep learning lies in its ability to capture interactions between features. For instance, we can incorporate environmental exposure, biological processes, and disease progression into different hierarchical levels of a model. By assessing neuron activations among these levels, we can identify nonlinear interactions between environmental exposures and biological effects at various levels. Computational perturbation can also be used to evaluate potential interactions, such as perturbing environmental exposure feature embeddings and observing the impact on biological effect feature embeddings to identify targets of environmental exposures. This approach has been applied to the discovery of gene–gene interactions, (11) though it requires significant computational resources. It is worth noting that perturbing two or more environmental exposure features simultaneously to calculate their combined impact on disease or biological effects could potentially evaluate the possible mixed effects of environmental exposures. Additionally, integrating laboratory data with models through active learning and carefully designed querying strategies can focus on the most uncertain or influential unlabeled samples for validation, reducing annotation costs and enhancing model interpretability. Integrating multimodal data. Currently, AI applications in HRA often focus on solving single tasks using one type of data. The future direction lies in integrating multimodal data to systematically determine how environmental exposures induce disease through multilevel biological processes. Given the increasing volume of multimodal data, there is an urgent need to develop suitable frameworks that align and fuse structured and unstructured modalities to generate accurate feature representations while preserving each modality’s biological information. One challenge in integrating exposome data is that different modalities often have varying resolutions and spatiotemporal scales. For example, chemical measurements in biological samples and gene expression or metabolite data reflect exposure only during specific time windows. In contrast, health outcomes, such as the onset or progression of chronic diseases, may not align with these time frames, as they often develop over longer periods. Another issue is that multimodal data often suffer from missing samples in one or more modalities. Because multimodal data complement each other, known modalities can be used to generate and impute missing features in others, improving the completeness of the data. Furthermore, the use of knowledge graph-based methods for multimodal data integration is still underexplored. KG can provide deeper biological insights for exposome data fusion, but this approach is still in its early stages and requires further development and optimization of graph construction and reasoning algorithms. As a result, the development of high-quality exposome data-sharing platforms has become urgent, as quality data are essential for effective modeling. The collection, cleaning, annotation, and integration of various datasets related to HRA face numerous challenges. While there are several large, high-quality databases, a significant amount of data remains dispersed across the literature, with inconsistencies in data sources, formats, and sample sizes. Therefore, the integration and standardization of data are crucial for creating high-quality open data-sharing platforms, such as ExposomeX and TOXRIC, to facilitate data growth. Constructing appropriate AI-Based models with stronger generalization ability. Before the selection of an algorithm, it is essential to adopt various strategies to address the challenges of small-sample sizes. Even the best models cannot derive valuable insights from inadequate data. Key strategies include actively collecting and curating diverse sample data (e.g., wearable device data and continuous environmental monitoring data), with efforts to maximize standardization and automation, thoroughly evaluating the distribution of small-sample groups to avoid biases, leveraging biological and environmental knowledge to guide feature engineering, and eliminating noise and outliers while appropriately handling missing data. When the appropriate model algorithm is being chosen, factors beyond accuracy must also be considered, such as model complexity, robustness, generalizability, and interpretability. A more systematic evaluation is required to ensure these aspects are balanced. In the context of transfer learning using large human biomonitoring databases, it is also important to account for potential biases arising from population selection. Furthermore, designing sophisticated models that incorporate existing prior biological knowledge is a promising approach. After model development, it is essential to assess the representativeness of small-sample size in HRA and consider the risks introduced by various biases. Bin Wang is a tenured Associate Professor and Vice Dean at the Institute of Reproductive and Child Health, Peking University. He also serves as an adjunct professor at the College of Urban and Environmental Sciences, Peking University. His primary research focuses on exposomics and AI-driven environmental health risk assessment. In collaboration with Prof. Mingliang Fang from Fudan University, he co-developed the integrative exposomics platform ExposomeX (www.exposomex.cn), accelerating research into the “Exposure–Biology–Disease” nexus. Prof. Wang has made significant contributions to the field by constructing statistical models to predict levels of common pollutants in the human body across specific regions. He has quantitatively assessed the links between pollution exposure in pregnant women from high-pollution areas and adverse reproductive health outcomes, providing critical evidence on the impacts of environmental pollutants on reproductive health. He is a pioneer in education, offering the course “Exposomics” to undergraduate and graduate students, as well as teaching in the prestigious international master’s program in global health and public health, “Environment & Health”. The authors thank the working group of environmental exposure and human health of the China Cohort Consortium (http://chinacohort.bjmu.edu.cn/). This study was supported by the National Natural Science Foundation of China (Grant 42477455), and the Strategy Priority Research Program (Category B) of the Chinese Academy of Sciences (XDB0750300). This article references 19 other publications. This article has not yet been cited by other publications.
下一代人类健康风险评估的小样本学习:利用人工智能、暴露数据和系统生物学
图1所示。环境暴露人体健康风险评估(HRA)小样本学习示意图。为了方便相关的HRA建模,需要考虑三个方面:研究设计、大数据收集和人工智能。未来模型的发展应该优先考虑平衡准确性和可解释性,有效地集成多模态数据,并构建强大的、可推广的人工智能模型,在解决数据偏差的同时结合复杂性和先前的生物学知识。利用大规模和可靠的人体生物监测数据库可以提供关于环境暴露与健康结果之间关系的先验知识。在过去的几十年里,在各个国家和地区建立了几项大规模的人体生物监测研究,对导致重大疾病的生活方式、环境和遗传因素进行流行病学研究。其中许多研究致力于开放数据和资源共享,以公平透明的方式提供数据。这些项目包括英国生物银行(UKB)、欧洲人类生物监测(HBM4 EU)、国家健康与营养检查调查(NHANES)、人类健康暴露分析资源(HHEAR)、中国健康与退休纵向研究(CHARLS)、环境对儿童健康结果的影响(ECHO)等。这些数据库提供了有关生物标本、遗传学、成像、医疗健康记录、生活方式、空气污染和化学品暴露的全面数据,为早期职业研究人员探索和测试有关环境暴露对人类疾病影响的假设提供了宝贵的资源。例如,Argentieri及其同事利用UKB的蛋白质组学数据(N = 45 441)开发了一种生物年龄预测模型,从2897种蛋白质中鉴定出204种关键血浆蛋白,用于构建蛋白质组年龄时钟。(5)来自中国(N = 3977)和芬兰(N = 1990)的独立队列验证,基于相同的204种蛋白质,显示出较高的预测准确性。通过利用在大人群中发现的具有强通用性的生物标志物,该方法有助于在较小的队列中进行准确的风险评估,增强对弱势群体的识别,并有助于减少错误,同时提高小人群中结果的可解释性。在缺乏人体生物监测数据的情况下,体外到体内外推法(IVIVE)也能够在个体水平上对小群体进行精确的健康风险预测。它还为常规使用化学品的优先次序、危害评估和监管决策提供了信心。最近,Han等人系统地总结和讨论了下一代HRA中的IVIVE方法,并从两个方面创新性地提出了前景。(6)一是扩大IVIVE的范围,如关注母体化合物及其代谢物的联合风险;二是在IVIVE中整合系统生物学、多组学、不良后果网络等新技术,实现更微观、更机械、更全面的风险评估。将多水平组学数据纳入小样本HRA可以更全面和准确地了解环境暴露如何影响健康。众所周知,环境暴露通常在不同程度上影响生物过程,导致不利的健康结果。然而,多组学研究的一个限制是成本高,单细胞转录组学的费用从每人1500美元到2000美元不等,导致高通量组学评估的典型小样本量。“维度诅咒”是多组学数据中一个重要的建模挑战。随着数据维数的增加,数据空间的稀疏度呈指数级增长,导致某些特征组合的发生概率变得极低,甚至完全看不见。这种稀疏性使得模型难以学习有意义的模式,从而影响了它们的泛化能力。通过不同测量技术获得的多组学数据往往表现出不同的特征和分布,这使这一挑战更加复杂。(7)面对小样本和高维的挑战,深度学习算法可以捕获复杂的非线性关系,并在进行降维的同时自动从低级组学数据中学习高质量的特征表示。例如,Cao等人开发了一个名为GLUE的模块化框架,该框架利用基于知识的图来模拟跨层调节交互,将组学层之间的特征空间连接起来。在该图中,顶点对应于不同组学层的特征,边缘表示它们之间的调节相互作用。 (8)该模型用于整合未配对的单细胞多组学数据,同时推断调控相互作用,在小样本情况下显示出出色的鲁棒性。为了缓解多组学数据中的多重共线性问题,必须保证数据的高质量和多样性,选择合适的模型训练和正则化策略,并整合现有的生物学知识(如基因调控网络和蛋白质-蛋白质相互作用)来提高模型效率。数据增加。这是一种用于生成与真实数据非常相似的新人工数据的技术,从而增强数据集。在这种情况下,ai生成内容(AIGC)利用大量未标记的数据来学习数据分布并生成样本,促进图像、文本、音频和视频的创建,同时在有限的标记数据条件下增强模型泛化能力。常见的模型包括生成对抗网络(GANs),它通过生成器和鉴别器之间的对抗训练生成真实样本,利用潜在空间的灵活性来有效捕获数据特征。扩散模型通过逐步去噪过程从随机噪声中生成高质量的数据,允许有效利用有限的数据并产生多样化的输出。变分自编码器(VAEs)通过编码器将数据压缩到潜在空间,然后解码生成新样本,在小样本学习场景中提供稳定的生成过程,保持数据的多样性和结构完整性。AIGCs在分子性质预测(9)和药物设计(10)方面取得了重大进展,但在与HRA相关的数据集中仍未得到充分的探索。转移学习。机器学习或深度学习模型可以在大型通用数据集上进行训练,并在下游任务中使用较小的、特定领域的数据集进行微调。迁移学习在HAR研究中的关键应用之一是利用化学物质的结构-性质关系和疾病基础的共享生物网络的见解,将传统污染物的知识应用于新出现的污染物,从常见疾病应用于罕见疾病。在应用迁移学习时,有必要在源人群和目标人群之间调整关键因素,包括暴露特征(例如,剂量-反应关系)、人群结构、疾病发病率和疾病类型。微调在迁移学习中是至关重要的,包括调整特定层的学习率,冻结早期层而调整后期层,以及使用数据增强或正则化来防止过拟合等策略。生成式预训练(GPT)利用注意机制和无监督预训练,已成为自然语言处理任务的先驱模型。受GPT的启发,研究人员将自我监督预训练应用于大规模单细胞转录组学数据,以开发可应用于较小患者数据集的基础模型,帮助确定疾病的候选治疗靶点。(11)在不久的将来,类似ChatGPT的模型可能会在人体生物监测数据库上进行训练,但需要进一步的研究来验证这些应用。多模式学习。在HRA研究中,暴露数据模式主要包括数字载体(例如,多组学数据和污染物浓度)、图形(例如,分子图形和生物知识图形)、文本(例如,电子健康记录和蛋白质序列)、视觉数据(例如,成像和遥感)和音频数据(例如,环境噪声)。在涉及系统生物学的任务中,多模式学习整合了来自各种模式的互补信息,提高了小样本学习下的表现,并为环境健康问题提供了更全面的视角。(12)多模态学习是一个通用的概念,可以通过各种架构来解决。一种常见的策略是对来自不同模态的数据进行联合建模,方法是为每种模态设计专门的体系结构,或者开发一个基础模型,将模态之间的相似概念映射到共享的潜在空间。这种方法能够为相同的语义或概念生成统一的内部表示(例如,遥感图像、传感器曝光数据和曝光事件描述的统一),并根据需要输出特定于任务的结果。基础模型已经成功地用于整合单细胞组学数据(8)和生物医学数据。(13)另一种方法是在模态之间构建知识图(knowledge graph, KGs)。 在系统生物学中,多层特征可以表示为异构图,捕获化学物质、基因、蛋白质和疾病之间的关联,图神经网络用于揭示这些实体之间的未知关系。多模态模型还为暴露提供了一种新的方法,将外部环境暴露因素与内部暴露生物标志物相结合,创建统一的特征向量,从而提供全面的个体暴露概况。稀疏结构的网络架构或算法。这些都是在生物信息的基础上设计的,可以减少模型参数。(14)生物信息设计是指模拟特定生物过程的神经网络中的隐藏节点,如DCell(细胞过程和功能)(15)和P-NET(基因和途径)。(14)更精简的架构和稀疏的网络结构可以提高小样本学习的准确性,同时量化网络中生物过程的相对重要性也增强了可解释性。深度森林模型(DF)是一种基于多层树的森林级联结构,适用于不同规模的数据集,超参数少,模型复杂度自适应生成。DF的模型复杂度可以在充分训练的情况下自适应确定,适用于小尺度的数据集。(16)受不利结果途径(AOP)框架的启发,我们可以将环境暴露、分子启动事件(MIEs)、关键分子事件(KEs)和不利结果整合到网络架构中,按复杂度排序。(17)值得注意的是,在具体的环境小样本研究中,这些方法不是单独进行的,而是经常系统地采用多种策略的组合。例如,Huang等人整合了多模态数据,包括基因组数据、细胞信号、基因表达水平和临床记录,构建了生物医学知识图谱,并进行了自我监督的预训练。(18)通过结合多模式学习和迁移学习的概念,他们的模型能够在严格的零注射条件下准确预测各种疾病的药物适应症和禁忌症,包括目前没有治疗药物可用的疾病。平衡模型的准确性和可解释性。虽然深度学习在各个领域取得了显著的成功,但可解释性仍然是一个反复出现的问题。在HRA的背景下,将模型权重参数转化为有意义的生物信息尤为重要。这不仅为医疗保健提供者提供了可靠的信息,而且为疾病进展提供了潜在的生物学解释。常见的方法包括SHAP (Shapley加性解释)和变压器架构中的注意力权重。然而,在小样本情况下,数据驱动的可解释性挖掘有时会产生虚假的重要特征,导致解释偏离生物事实和深入的环境毒理学研究。(19)另一种方法是通过计算扰动特征(例如,删除、随机屏蔽或组学数据反转)来评估与疾病相关的关键环境暴露和生物学特征,并量化这些扰动对预测结果的影响。为了提高可解释性,干扰已知特征(例如,MIE和KE)而不是随机特征可能是有用的,尽管这需要与毒性途径相关的先验知识。虽然上面描述的方法适用于评估单个特征的重要性,但深度学习的优势在于它能够捕获特征之间的交互。例如,我们可以将环境暴露、生物过程和疾病进展纳入模型的不同层次。通过评估这些水平之间的神经元激活,我们可以识别不同水平的环境暴露与生物效应之间的非线性相互作用。计算摄动也可用于评估潜在的相互作用,如干扰环境暴露特征嵌入和观察对生物效应特征嵌入的影响,以确定环境暴露的目标。虽然这种方法需要大量的计算资源,但它已被应用于发现基因-基因相互作用(11)。值得注意的是,同时干扰两种或两种以上的环境暴露特征,以计算它们对疾病或生物效应的综合影响,可能会评估环境暴露可能产生的混合效应。 此外,通过主动学习和精心设计的查询策略将实验室数据与模型集成,可以关注最不确定或最具影响力的未标记样本进行验证,从而降低注释成本并增强模型的可解释性。整合多模态数据。目前,人工智能在人力资源管理中的应用往往侧重于使用一种类型的数据来解决单一任务。未来的方向在于整合多模态数据,系统地确定环境暴露如何通过多层次的生物过程诱发疾病。鉴于多模态数据量的增加,迫切需要开发合适的框架来对齐和融合结构化和非结构化模态,以生成准确的特征表示,同时保留每种模态的生物信息。整合暴露数据的一个挑战是,不同的模式往往具有不同的分辨率和时空尺度。例如,生物样品中的化学测量和基因表达或代谢物数据仅反映特定时间窗内的暴露。相比之下,健康结果,如慢性病的发病或进展,可能与这些时间框架不一致,因为它们往往需要更长的时间才能发展。另一个问题是,多模态数据经常在一个或多个模态中丢失样本。由于多模态数据是相互补充的,已知的模态可以用来生成和推断其他模态的缺失特征,从而提高数据的完整性。此外,使用基于知识图的方法进行多模态数据集成的研究还不够充分。KG可以为暴露数据融合提供更深入的生物学见解,但这种方法仍处于早期阶段,需要进一步开发和优化图构建和推理算法。因此,开发高质量的暴露数据共享平台变得迫在眉睫,因为高质量的数据是有效建模的必要条件。与HRA相关的各种数据集的收集、清理、注释和集成面临着许多挑战。虽然有几个大型、高质量的数据库,但大量的数据仍然分散在文献中,数据源、格式和样本量不一致。因此,数据的整合和标准化对于创建高质量的开放数据共享平台至关重要,例如ExposomeX和TOXRIC,以促进数据的增长。构建合适的、泛化能力更强的人工智能模型。在选择算法之前,必须采用各种策略来解决小样本量的挑战。即使是最好的模型也无法从不充分的数据中得出有价值的见解。关键策略包括积极收集和整理各种样本数据(例如,可穿戴设备数据和连续环境监测数据),努力最大限度地实现标准化和自动化,彻底评估小样本组的分布以避免偏差,利用生物和环境知识指导特征工程,在适当处理缺失数据的同时消除噪声和异常值。在选择合适的模型算法时,还必须考虑精度以外的因素,如模型复杂性、鲁棒性、泛化性和可解释性。需要进行更系统的评价,以确保这些方面得到平衡。在使用大型人类生物监测数据库进行迁移学习的背景下,考虑群体选择产生的潜在偏差也很重要。此外,设计包含现有先验生物学知识的复杂模型是一种很有前途的方法。在模型建立之后,评估小样本量在HRA中的代表性和考虑各种偏差带来的风险是至关重要的。王斌是北京大学生殖与儿童健康研究所的终身副教授和副院长。他也是北京大学城市与环境科学学院的兼职教授。他的主要研究方向是暴露学和人工智能驱动的环境健康风险评估。他与复旦大学方明亮教授合作,共同开发了综合暴露学平台ExposomeX (www.exposomex.cn),加速了“暴露-生物学-疾病”关系的研究。王教授通过构建统计模型来预测人体特定区域的常见污染物水平,在该领域做出了重大贡献。他定量评估了来自高污染地区的孕妇接触污染与不利生殖健康结果之间的联系,就环境污染物对生殖健康的影响提供了重要证据。 他是教育领域的先驱,为本科生和研究生开设了“暴露学”课程,并在全球卫生和公共卫生领域享有盛誉的国际硕士课程“环境与公共卫生”中任教。健康。”作者感谢中国队列联合会环境暴露与人类健康工作组(http://chinacohort.bjmu.edu.cn/)。本研究得到国家自然科学基金项目(no . 42477455)和中国科学院战略重点研究项目(no . XDB0750300)的支持。本文引用了其他19个出版物。这篇文章尚未被其他出版物引用。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

环境科学与技术
环境科学-工程:环境
CiteScore
17.50
自引率
9.60%
发文量
12359
审稿时长
2.8 months
期刊介绍:
Environmental Science & Technology (ES&T) is a co-sponsored academic and technical magazine by the Hubei Provincial Environmental Protection Bureau and the Hubei Provincial Academy of Environmental Sciences.
Environmental Science & Technology (ES&T) holds the status of Chinese core journals, scientific papers source journals of China, Chinese Science Citation Database source journals, and Chinese Academic Journal Comprehensive Evaluation Database source journals. This publication focuses on the academic field of environmental protection, featuring articles related to environmental protection and technical advancements.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


