FBP1 controls liver cancer evolution from senescent MASH hepatocytes
IF 50.5
1区 综合性期刊
Q1 MULTIDISCIPLINARY SCIENCES
引用次数: 0
Abstract
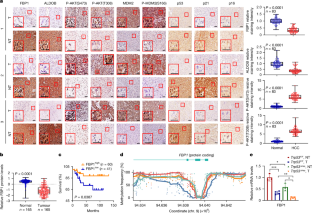
Hepatocellular carcinoma (HCC) originates from differentiated hepatocytes undergoing compensatory proliferation in livers damaged by viruses or metabolic-dysfunction-associated steatohepatitis (MASH)1. While increasing HCC risk2, MASH triggers p53-dependent hepatocyte senescence3, which we found to parallel hypernutrition-induced DNA breaks. How this tumour-suppressive response is bypassed to license oncogenic mutagenesis and enable HCC evolution was previously unclear. Here we identified the gluconeogenic enzyme fructose-1,6-bisphosphatase 1 (FBP1) as a p53 target that is elevated in senescent-like MASH hepatocytes but suppressed through promoter hypermethylation and proteasomal degradation in most human HCCs. FBP1 first declines in metabolically stressed premalignant disease-associated hepatocytes and HCC progenitor cells4,5, paralleling the protumorigenic activation of AKT and NRF2. By accelerating FBP1 and p53 degradation, AKT and NRF2 enhance the proliferation and metabolic activity of previously senescent HCC progenitors. The senescence-reversing and proliferation-supportive NRF2–FBP1–AKT–p53 metabolic switch, operative in mice and humans, also enhances the accumulation of DNA-damage-induced somatic mutations needed for MASH-to-HCC progression. The p53 target FBP1 is elevated in senescent-like metabolic-dysfunction-associated steatohepatitis hepatocytes but suppressed through promoter hypermethylation and proteasomal degradation in most human hepatocellular carcinomas.

FBP1控制衰老的MASH肝细胞的肝癌进化
肝细胞癌(HCC)起源于分化的肝细胞在被病毒或代谢功能障碍相关的脂肪性肝炎(MASH)损伤的肝脏中进行代偿性增殖1。在增加HCC风险的同时,MASH会触发p53依赖性肝细胞衰老,我们发现这与高营养诱导的DNA断裂相似。这种肿瘤抑制反应是如何被绕过以允许致癌突变并使HCC进化的,这在以前是不清楚的。在这里,我们发现糖异生酶果糖-1,6-二磷酸酶1 (FBP1)是p53的靶标,在衰老样MASH肝细胞中升高,但在大多数人类hcc中通过启动子超甲基化和蛋白酶体降解抑制。FBP1首先在代谢应激的癌前疾病相关肝细胞和HCC祖细胞中下降4,5,与AKT和NRF2的致瘤活性相似。通过加速FBP1和p53的降解,AKT和NRF2增强了先前衰老的HCC祖细胞的增殖和代谢活性。逆转衰老和支持增殖的NRF2-FBP1-AKT-p53代谢开关在小鼠和人类中起作用,也促进了dna损伤诱导的体细胞突变的积累,这是mash向hcc进展所必需的。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Nature
综合性期刊-综合性期刊
CiteScore
90.00
自引率
1.20%
发文量
3652
审稿时长
3 months
期刊介绍:
Nature is a prestigious international journal that publishes peer-reviewed research in various scientific and technological fields. The selection of articles is based on criteria such as originality, importance, interdisciplinary relevance, timeliness, accessibility, elegance, and surprising conclusions. In addition to showcasing significant scientific advances, Nature delivers rapid, authoritative, insightful news, and interpretation of current and upcoming trends impacting science, scientists, and the broader public. The journal serves a dual purpose: firstly, to promptly share noteworthy scientific advances and foster discussions among scientists, and secondly, to ensure the swift dissemination of scientific results globally, emphasizing their significance for knowledge, culture, and daily life.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


