Multiconfigurational Electronic Structure of Nickel Cross-Coupling Catalysts Revealed by X-ray Absorption Spectroscopy
IF 4.6
2区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
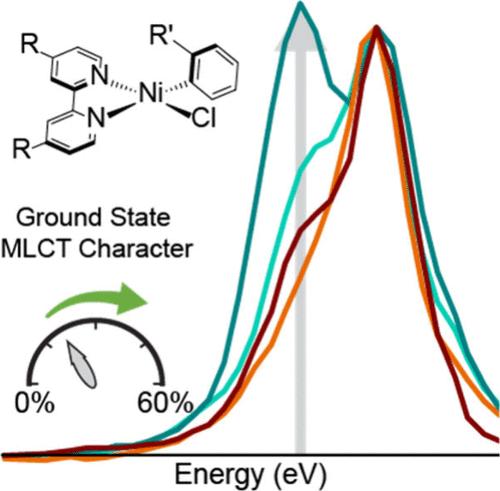
NiII 2,2′-bipyridine complexes are commonly invoked intermediates in metallaphotoredox cross-coupling reactions. Despite their ubiquity, design principles targeting improved catalytic performance remain underdetermined. A series of Ni(Rbpy)(R′Ar)Cl (R = MeOOC, t-Bu, R′ = CH3, CF3) complexes were proposed to have multiconfigurational electronic structures on the basis of multiconfigurational/multireference calculations, with significant mixing of Ni → bpy metal-to-ligand charge transfer (MLCT) configurations into the ground-state wave function. Here, Ni K-edge and L2,3-edge X-ray absorption spectroscopies provide experimental support for the highly covalent and multiconfigurational electronic structures of these complexes. The pre-edge intensity in the K-edge spectrum reflects highly covalent Ni–aryl bonding. The L3-edge spectral shape is dependent on ligand functionalization, and a feature reflecting the MLCT character is assigned using prior ab initio and new semiempirical calculations. The results suggest the push/pull effects of the aryl/bpy ligands moderate the changes in electron density on Ni during the multiredox cross-coupling reaction cycle.
用x射线吸收光谱研究镍交叉偶联催化剂的多构型电子结构
NiII 2,2′-联吡啶配合物是金属氧化还原交叉偶联反应中常用的中间体。尽管它们无处不在,但旨在提高催化性能的设计原则仍未确定。根据多配位/多参量计算,一系列 Ni(Rbpy)(R′Ar)Cl(R = MeOOC、t-Bu,R′ = CH3、CF3)复合物被认为具有多配位电子结构,在基态波函数中显著混合了 Ni → bpy 金属配体电荷转移(MLCT)构型。在这里,Ni K 边和 L2,3 边 X 射线吸收光谱为这些复合物的高共价和多构型电子结构提供了实验支持。K 边光谱中的前边强度反映了高度共价的镍芳基键。L3 边谱的形状取决于配体的官能化程度,利用先前的 ab initio 计算和新的半经验计算,确定了反映 MLCT 特性的特征。结果表明,在多氧化还原交叉偶联反应周期中,芳基/苄基配体的推/拉效应缓和了镍上电子密度的变化。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

The Journal of Physical Chemistry Letters
CHEMISTRY, PHYSICAL-NANOSCIENCE & NANOTECHNOLOGY
CiteScore
9.60
自引率
7.00%
发文量
1519
审稿时长
1.6 months
期刊介绍:
The Journal of Physical Chemistry (JPC) Letters is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, chemical physicists, physicists, material scientists, and engineers. An important criterion for acceptance is that the paper reports a significant scientific advance and/or physical insight such that rapid publication is essential. Two issues of JPC Letters are published each month.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


