Irshad Ul Haq, Peter Kennedy, Kathryn M. Schreiner, Julia C. Agnich, Jonathan S. Schilling
{"title":"Gene Expression by a Model Fungus in the Ascomycota Provides Insight Into the Decay of Fungal Necromass","authors":"Irshad Ul Haq, Peter Kennedy, Kathryn M. Schreiner, Julia C. Agnich, Jonathan S. Schilling","doi":"10.1111/1462-2920.70006","DOIUrl":null,"url":null,"abstract":"<p>Dead fungal cells, known as necromass, are increasingly recognised as significant contributors to long-term soil carbon pools, yet the genes involved in necromass decomposition are poorly understood. In particular, how microorganisms degrade necromass with differing initial cell wall chemical compositions using carbohydrate-active enzymes (CAZymes) has not been well studied. Based on the frequent occurrence and high abundance of the fungal genus <i>Trichoderma</i> on decaying fungal necromass in situ, we grew <i>Trichoderma reesei</i> RUT-C30 on low and high melanin necromass of <i>Hyaloscypha bicolor</i> (Ascomycota) in liquid cultures and assessed <i>T. reesei</i> gene expression relative to each other and relative to glucose. Transcriptome data revealed that <i>T. reesei</i> up-regulated many genes (over 100; necromass versus glucose substrate) coding for CAZymes, including enzymes that would target individual layers of an Ascomycota fungal cell wall. We also observed differential expression of protease- and laccase-encoding genes on high versus low melanin necromass, highlighting a subset of genes (fewer than 15) possibly linked to the deconstruction of melanin, a cell wall constituent that limits necromass decay rates in nature. Collectively, these results advance our understanding of the genomic traits underpinning the rates and fates of carbon turnover in an understudied pool of Earth's belowground carbon, fungal necromass.</p>","PeriodicalId":11898,"journal":{"name":"Environmental microbiology","volume":"26 12","pages":""},"PeriodicalIF":4.3000,"publicationDate":"2024-12-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1111/1462-2920.70006","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Environmental microbiology","FirstCategoryId":"99","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/1462-2920.70006","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"MICROBIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
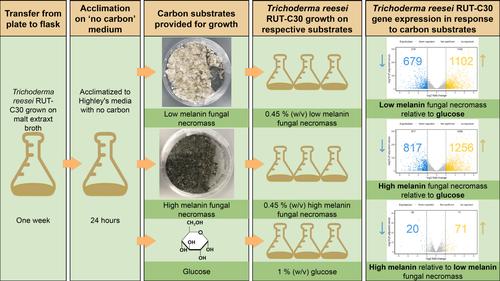
Dead fungal cells, known as necromass, are increasingly recognised as significant contributors to long-term soil carbon pools, yet the genes involved in necromass decomposition are poorly understood. In particular, how microorganisms degrade necromass with differing initial cell wall chemical compositions using carbohydrate-active enzymes (CAZymes) has not been well studied. Based on the frequent occurrence and high abundance of the fungal genus Trichoderma on decaying fungal necromass in situ, we grew Trichoderma reesei RUT-C30 on low and high melanin necromass of Hyaloscypha bicolor (Ascomycota) in liquid cultures and assessed T. reesei gene expression relative to each other and relative to glucose. Transcriptome data revealed that T. reesei up-regulated many genes (over 100; necromass versus glucose substrate) coding for CAZymes, including enzymes that would target individual layers of an Ascomycota fungal cell wall. We also observed differential expression of protease- and laccase-encoding genes on high versus low melanin necromass, highlighting a subset of genes (fewer than 15) possibly linked to the deconstruction of melanin, a cell wall constituent that limits necromass decay rates in nature. Collectively, these results advance our understanding of the genomic traits underpinning the rates and fates of carbon turnover in an understudied pool of Earth's belowground carbon, fungal necromass.
期刊介绍:
Environmental Microbiology provides a high profile vehicle for publication of the most innovative, original and rigorous research in the field. The scope of the Journal encompasses the diversity of current research on microbial processes in the environment, microbial communities, interactions and evolution and includes, but is not limited to, the following:
the structure, activities and communal behaviour of microbial communities
microbial community genetics and evolutionary processes
microbial symbioses, microbial interactions and interactions with plants, animals and abiotic factors
microbes in the tree of life, microbial diversification and evolution
population biology and clonal structure
microbial metabolic and structural diversity
microbial physiology, growth and survival
microbes and surfaces, adhesion and biofouling
responses to environmental signals and stress factors
modelling and theory development
pollution microbiology
extremophiles and life in extreme and unusual little-explored habitats
element cycles and biogeochemical processes, primary and secondary production
microbes in a changing world, microbially-influenced global changes
evolution and diversity of archaeal and bacterial viruses
new technological developments in microbial ecology and evolution, in particular for the study of activities of microbial communities, non-culturable microorganisms and emerging pathogens


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


