Ayoub Aouina, Pedro Borlido, Miguel A L Marques, Silvana Botti
{"title":"Assessing Exchange-Correlation Functionals for Accurate Densities of Solids.","authors":"Ayoub Aouina, Pedro Borlido, Miguel A L Marques, Silvana Botti","doi":"10.1021/acs.jctc.4c01042","DOIUrl":null,"url":null,"abstract":"<p><p>The success of Kohn-Sham density functional theory in predicting electronic properties from first-principles is key to its ubiquitous presence in condensed matter research. Central to this theory is the exchange-correlation functional, which can only be written in an approximate form using a handful of exact constraints. A recent criticism of these approximations is that they are designed to give an accurate description of the energy at the expense of a poor representation of the density, which is contrary to the spirit of density functional theory. These conclusions are drawn from studies of atoms or small molecules, where exact results are available. To shed light on this issue, we use the almost exact densities and energies of three prototypical solids (a semiconductor, silicon, an insulator, sodium chloride, and a metal, copper) to compare the performance of exchange-correlation functionals from all rungs of Jacob's ladder. By examining their errors in reproducing both energy and density, we show that several hybrids and semilocal functionals perform consistently well. Furthermore, functionals built to reproduce exact constraints tend to be among the top performers for all tested material classes, strengthening the argument for using these constraints in functional construction. On average, functionals published up to the early 2000s simultaneously improve the prediction of both densities and energies. This is often not the case for more recent functionals, although errors in energy and density continue to evolve in a correlated manner.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":" ","pages":"10852-10860"},"PeriodicalIF":5.5000,"publicationDate":"2024-12-24","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11672669/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.4c01042","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/12/3 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
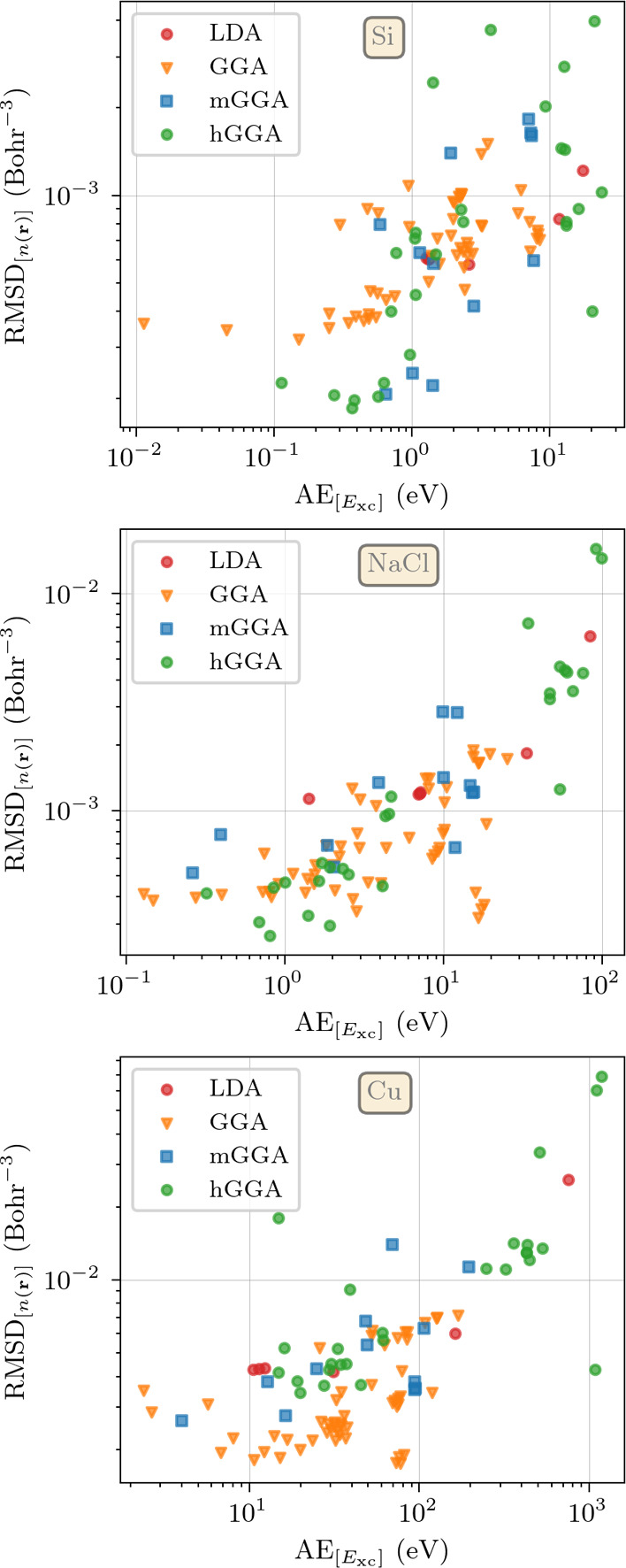
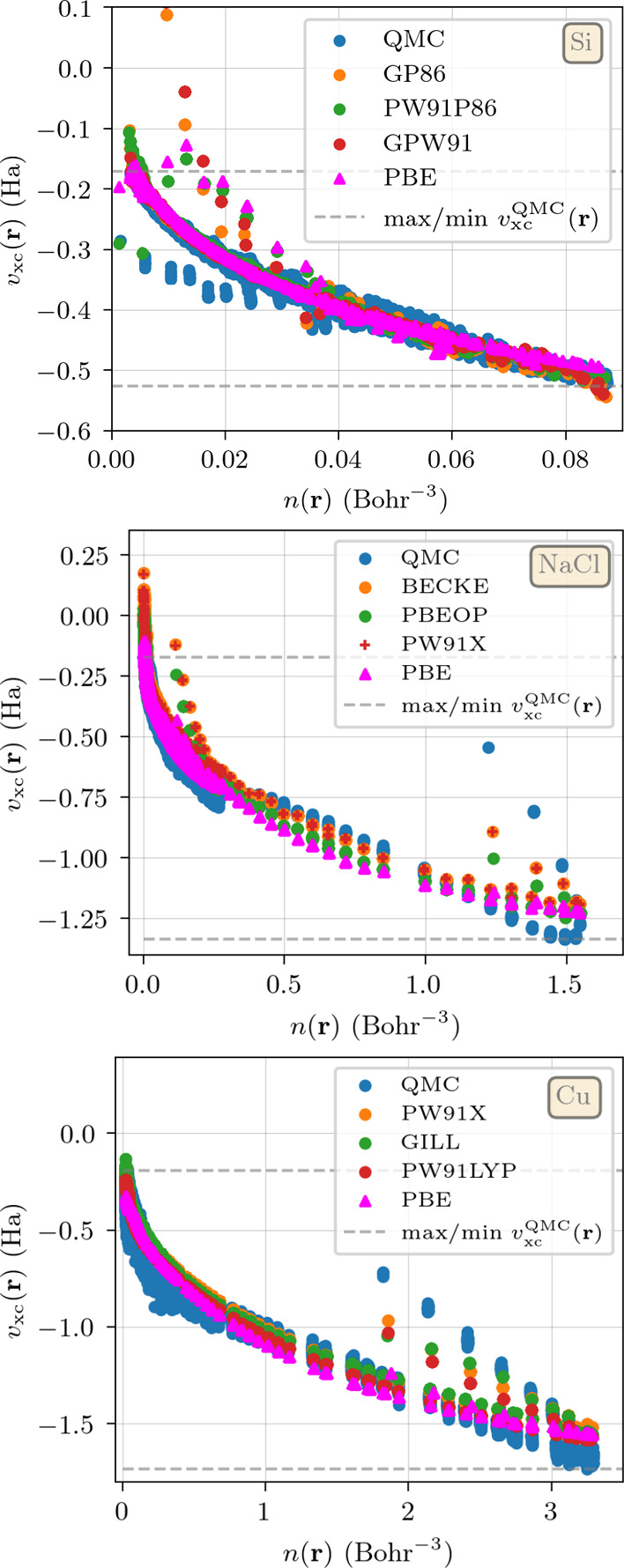
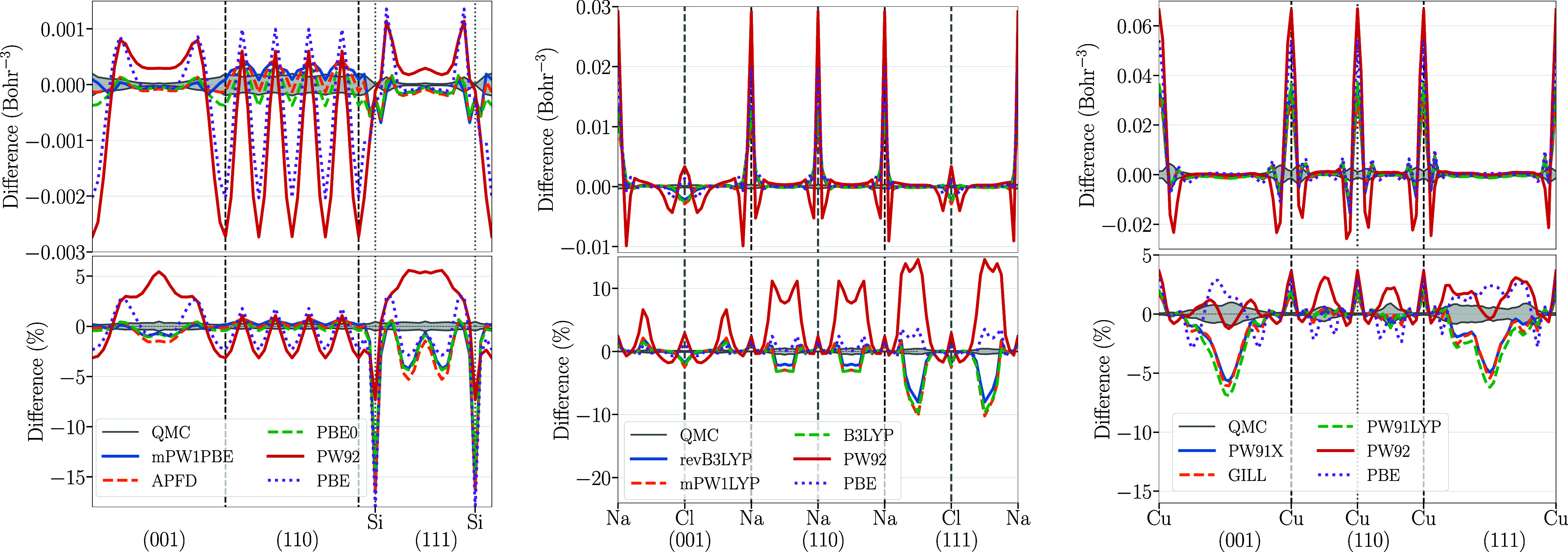
The success of Kohn-Sham density functional theory in predicting electronic properties from first-principles is key to its ubiquitous presence in condensed matter research. Central to this theory is the exchange-correlation functional, which can only be written in an approximate form using a handful of exact constraints. A recent criticism of these approximations is that they are designed to give an accurate description of the energy at the expense of a poor representation of the density, which is contrary to the spirit of density functional theory. These conclusions are drawn from studies of atoms or small molecules, where exact results are available. To shed light on this issue, we use the almost exact densities and energies of three prototypical solids (a semiconductor, silicon, an insulator, sodium chloride, and a metal, copper) to compare the performance of exchange-correlation functionals from all rungs of Jacob's ladder. By examining their errors in reproducing both energy and density, we show that several hybrids and semilocal functionals perform consistently well. Furthermore, functionals built to reproduce exact constraints tend to be among the top performers for all tested material classes, strengthening the argument for using these constraints in functional construction. On average, functionals published up to the early 2000s simultaneously improve the prediction of both densities and energies. This is often not the case for more recent functionals, although errors in energy and density continue to evolve in a correlated manner.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


