Mimmi Tang, Katrine Elisabeth Karmisholt, Martin Gillstedt, Jaishri O. Blakeley, Joshua Roberts, Jørgen Serup, Torsten Bove, Sirkku Peltonen
{"title":"Protocol for high-intensity focused ultrasound (HIFU) treatment of cutaneous neurofibromas","authors":"Mimmi Tang, Katrine Elisabeth Karmisholt, Martin Gillstedt, Jaishri O. Blakeley, Joshua Roberts, Jørgen Serup, Torsten Bove, Sirkku Peltonen","doi":"10.1002/jvc2.543","DOIUrl":null,"url":null,"abstract":"<p>High-intensity focused ultrasound (HIFU) is widely used for various applications including treatment of cancers of the thyroid,<span><sup>1</sup></span> prostate,<span><sup>2</sup></span> pancreas<span><sup>3</sup></span> and bone metastases<span><sup>4</sup></span> as well as targeting deep brain nuclei to treat tremor.<span><sup>5</sup></span> HIFU systems for these treatments operate at frequencies of 0.5–3 MHz. Ultrasound is also used for aesthetic applications, such as body contouring, at frequencies of 4–12 MHz.</p><p>The 20 MHz HIFU device used in this study is developed for selective destructive treatments of very small targets in the epidermis and the dermis while sparing the surroundings.<span><sup>6</sup></span> Each dose produces a combination of mechanical disruption and local heating to approximately 60–65°C at the focal point of the focused ultrasound beam emitted from a concave transducer. The primary effect is cell necrosis. This 20 MHz HIFU has previously been documented useful in actinic keratoses basal cell carcinoma, Kaposi sarcoma and superficial vascular lesions.<span><sup>7-9</sup></span></p><p>The present study reports on efforts to optimise the dose of HIFU applied to cutaneous neurofibroma (cNF) in patients with Neurofibromatosis Type 1 (NF1). This is a second phase extension of an original study of the tolerability and safety of HIFU.<span><sup>10</sup></span> The two studies are referred to as Phase A and Phase B, respectively. There are no currently approved therapies for NF1-associated cNFs. These skin tumours can count in the hundreds or thousands for a given individual. Due to their associated symptoms of itching, pain and disfigurement, cNF represents a major unmet need.</p><p>The study was a 6-month prospective open-label study conducted at the Department of Dermatology and Venereology, University of Gothenburg, Gothenburg, Sweden. The study was registered on www.clinicaltrials.gov under number NCT05119582. The original study included 20 participants across two centres, Bispebjerg Hospital in Copenhagen, Denmark and University of Gothenburg.<span><sup>10</sup></span> Seven patients from the original cohort were recruited for an additional dosimetry study.</p><p>Written informed consent with permission to use anonymized data and photos was obtained on study start in February 2023. All patients had clinically or otherwise verified NF1. cNFs were categorised into flat, sessile and globular types.</p><p>Tumours were numbered, and the locations of the included tumours were drawn on a plastic registration sheet to identify the treatment sites at follow-up visits. Tumours were documented by conventional 2D clinical photos at baseline, 1 week, 3 months and 6 months after treatment. A total of four to nine cNFs per patient were treated. An additional two cNFs were included as control tumours. Data were collected in case report forms on the Research Electronic Data Capture (RedCap) platform.</p><p>Each selected cNF received one treatment with the HIFU system (System ONE-M, TOOsonix A/S, Hoersholm, Denmark) (Figure 1). A handpiece with a nominal focal depth of 2.3 mm was used. The handpiece was positioned on each cNF to be treated. The exact positioning was displayed as a dermoscopic real-time video feed from a camera housed in the handpiece. The dosing was set to duration 500 ms and acoustic output power of 1.8 W, resulting in a dose energy of 0.9 J/dose. Doses were applied every 1–3 s (video), with approximately 1 mm spacing between each dose to fully cover the tumour with a margin of about 1 mm in the perilesional skin. Local anaesthesia was not used.</p><p>Adverse events of the treatment were assessed using six-point grading scales according to the CTCAE system (US Department of Health and Human Services 2017). Patient-reported outcome and patient-reported experience were measured using Likert scales with rating from 0 to 10 or 5-point rating scales of effects (e.g., ranging from absent effect or very satisfied to severe effect or very dissatisfied). To compare the pain in Phase A and Phase B, 2-sample <i>t</i> test was carried out. To estimate the efficacy of treatment, each tumour was evaluated clinically using a 0–10-point scale where 5 was the neutral baseline, 1 major increase in size of the cNF and 10 complete disappearance of the tumour.</p><p>The statistical analysis was conducted using The R Foundation for Statistical Computing, Vienna, Austria, http://www.Rproject.org software. Quantitative study variables were described as mean ± SD and qualitative binary variables as <i>n</i> (%). Wilcoxon's rank-sum test was used for two-sample comparisons. Pearson's correlation test was used to analyse relationships between erosions after the treatment and efficacy at 6 months. All tests were two-sided and <i>p</i> < 0.05 was considered as statistically significant.</p><p>The primary endpoints were safety and tolerability of the HIFU treatment. The secondary endpoints were other registered biological responses after the treatment, feasibility of the device, change in cNF size and patient's assessment of satisfaction with the treatment and the device.</p><p>Seven patients and a total of 68 tumours 2–5 mm in width (median diameter 5 mm) located on the anterior or posterior trunk or on the arms were included in the study. Demographics of the patients and characteristics of the included tumours are shown in Table 1. Of all tumours included, 54 were treated and 14 were untreated controls. The number of doses per lesion varied from 13 to 25; median 18. All enrolled participants completed all required study evaluations.</p><p>As observed in the Phase A study, the treatment resulted in mild, local treatment effects.<span><sup>10</sup></span> Local immediate wheal and flare reactions and oedema were either absent or mild, and no bruising was observed. Experience of pain during the treatment was reported by patients on a 0–10 scale and was graded higher than in Phase A (median 5 <i>vs</i>. 3.5 in Phase A), but without a statistically significant difference (<i>p</i> = 0.400). Pain was instant and disappeared immediately after the dose. At the 6-month follow-up visit, 6/7 participants reported no side effects, and one had mild side effects only. There were no instrument-related adverse events. The primary endpoints concerning safety and tolerability were thus fulfilled.</p><p>On follow-up visits, superficial erosions were seen in 24/54 (44%) of tumours during the first week after the treatment. All erosions healed in 1–2 weeks and none developed scars. Pigment changes after 6 months were noted in 10/54 (18.5%) of tumours; all being tumours with erosion after 1 week. Pigment variation with hypopigmentation in the middle surrounded by a hyperpigmented rim was encountered only on skin phototype IV. Summary of observations of pigmentation is included in Table 2. An example of the various biological responses of three cNFs located close to each other in a patient with phototype IV is shown in Figure 2.</p><p>The analysis of efficacy 6 months after a single treatment of each cNF as measured using a ruler demonstrated that 70% of the treated cNFs showed at least some reduction in size. Major reduction or complete disappearance was observed in 26% of the tumours (Table 3). The efficacy in Phase B with a median of 6 was lower than the median of 7 in Phase A, but the difference was outside statistical significance (<i>p</i> = 0.09). Further statistical analysis of the data showed significant correlation between reduction in tumour size and observation of a superficial erosion 1 week after treatment (correlation coefficient 0.449, <i>p</i> = 0,005).</p><p>This study sought to optimise the dosing of a HIFU treatment which would enable noninvasive therapy of large numbers of cNFs in one session. In Phase A, 250 ms/dose with 0.7 J/dose was used and showed major reduction in 49% of tumours. Although this was encouraging, we tried to further optimise the dosing in Phase B with a dose of 500 ms/dose with 0.9 J/dose. The treatment-related effects remained very mild. The settings used in Phase B did cause a higher level of immediate pain during the treatment. Efficacy as assessed by absolute cNF size tended to be lower in Phase B than Phase A. Interestingly, erosions at 1 week after treatment were strongly correlated with efficacy, indicating that settings in Phase B are preferable in some metrics. Erosions did lead to pigment changes which were most pronounced in patients with darker skin type. The erosions were not seen immediately after treatment but developed within a few days. However, no scarring was encountered.</p><p>The limitation of the present study is the small number of patients. That said, 54 cNFs were treated across multiple cNF subtypes and sizes and, therefore, the efficacy seen in this extension supports the tolerability and efficacy seen in the Phase A study. As a result of these data, the 20 MHz HIFU device received CE Mark approval for treatment of cNF in people with NF1 in the European Union. A guidance for use in this context generated from the Phase A and Phase B studies, and a video showing the treatment are provided as supplementary files.</p><p>In conclusion, 20 MHz HIFU is demonstrated as a tolerable, safe and efficient method for the treatment of smaller cNFs in patients with NF1. The method is minimally invasive and with potential for clinical use for field eradication of early-stage cNF in different anatomical sites. Given the lack of approved medicinal therapies for cNF as well as the safety and efficacy demonstrated across both the Phases A and B portions of the study, 20 MHz HIFU has been shown to be an important and viable treatment option for early-stage cNFs (Video 1).</p><p><i>Conceptualisation</i>: Sirkku Peltonen, Jørgen Serup, Jaishri O. Blakeley, Katrine Elisabeth Karmisholt and Torsten Bove. <i>Data curation</i>: Sirkku Peltonen, Katrine Elisabeth Karmisholt, Joshua Roberts, Martin Gillstedt and Mimmi Tang. <i>Formal analysis</i>: Sirkku Peltonen, Katrine Elisabeth Karmisholt, Martin Gillstedt, Torsten Bove and Mimmi Tang. <i>Funding acquisition</i>: Sirkku Peltonen, Jørgen Serup, Jaishri O. Blakeley, Torsten Bove and Katrine Elisabeth Karmisholt. <i>Investigation</i>: Sirkku Peltonen, Mimmi Tang and Torsten Bove. <i>Methodology</i>: Sirkku Peltonen, Jørgen Serup, Jaishri O. Blakeley, Torsten Bove, Katrine Elisabeth Karmisholt and Joshua Roberts. <i>Project administration</i>: Sirkku Peltonen, Jørgen Serup, Jaishri O. Blakeley, Joshua Roberts, Torsten Bove and Katrine Elisabeth Karmisholt. <i>Resources</i>: Sirkku Peltonen, Jørgen Serup, Mimmi Tang, Torsten Bove and Katrine Elisabeth Karmisholt. <i>Statistics</i>: Martin Gillstedt and Torsten Bove. <i>Supervision</i>: Sirkku Peltonen, Jørgen Serup, Jaishri O. Blakeley, Joshua Roberts and Katrine Elisabeth Karmisholt. <i>Validation</i>: Sirkku Peltonen, Jørgen Serup, Martin Gillstedt and Katrine Elisabeth Karmisholt. <i>Visualisation</i>: Sirkku Peltonen, Jørgen Serup, Martin Gillstedt, Mimmi Tang, Torsten Bove and Katrine Elisabeth Karmisholt. <i>Writing—Original draft preparation</i>: Sirkku Peltonen, Mimmi Tang and Torsten Bove. <i>Writing—Review, editing and final approval of the manuscript</i>: Sirkku Peltonen, Jørgen Serup, Mimmi Tang, Martin Gillstedt, Jaishri O.Blakeley, Joshua Roberts, Torsten Bove and Katrine Elisabeth Karmisholt.</p><p>JS is a sponsor for this clinical investigation funded by NTAP. JOB and JR are officers of NTAP. TB is a shareholder and CEO of TOOsonix A/S. The remaining authors declare no conflict of interest.</p><p>The study was approved by the Danish National Ethics Committee, the Swedish Ethics Authority and the Danish and Swedish Medical Agencies. All patients in this manuscript have given written informed consent for participation in the study and the use of their deidentified, anonymized, aggregated data and their case details (including photographs) for publication. The study followed the Good Clinical Practice requirements of ICH.</p>","PeriodicalId":94325,"journal":{"name":"JEADV clinical practice","volume":"3 5","pages":"1723-1729"},"PeriodicalIF":0.0000,"publicationDate":"2024-09-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jvc2.543","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"JEADV clinical practice","FirstCategoryId":"1085","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jvc2.543","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
High-intensity focused ultrasound (HIFU) is widely used for various applications including treatment of cancers of the thyroid,1 prostate,2 pancreas3 and bone metastases4 as well as targeting deep brain nuclei to treat tremor.5 HIFU systems for these treatments operate at frequencies of 0.5–3 MHz. Ultrasound is also used for aesthetic applications, such as body contouring, at frequencies of 4–12 MHz.
The 20 MHz HIFU device used in this study is developed for selective destructive treatments of very small targets in the epidermis and the dermis while sparing the surroundings.6 Each dose produces a combination of mechanical disruption and local heating to approximately 60–65°C at the focal point of the focused ultrasound beam emitted from a concave transducer. The primary effect is cell necrosis. This 20 MHz HIFU has previously been documented useful in actinic keratoses basal cell carcinoma, Kaposi sarcoma and superficial vascular lesions.7-9
The present study reports on efforts to optimise the dose of HIFU applied to cutaneous neurofibroma (cNF) in patients with Neurofibromatosis Type 1 (NF1). This is a second phase extension of an original study of the tolerability and safety of HIFU.10 The two studies are referred to as Phase A and Phase B, respectively. There are no currently approved therapies for NF1-associated cNFs. These skin tumours can count in the hundreds or thousands for a given individual. Due to their associated symptoms of itching, pain and disfigurement, cNF represents a major unmet need.
The study was a 6-month prospective open-label study conducted at the Department of Dermatology and Venereology, University of Gothenburg, Gothenburg, Sweden. The study was registered on www.clinicaltrials.gov under number NCT05119582. The original study included 20 participants across two centres, Bispebjerg Hospital in Copenhagen, Denmark and University of Gothenburg.10 Seven patients from the original cohort were recruited for an additional dosimetry study.
Written informed consent with permission to use anonymized data and photos was obtained on study start in February 2023. All patients had clinically or otherwise verified NF1. cNFs were categorised into flat, sessile and globular types.
Tumours were numbered, and the locations of the included tumours were drawn on a plastic registration sheet to identify the treatment sites at follow-up visits. Tumours were documented by conventional 2D clinical photos at baseline, 1 week, 3 months and 6 months after treatment. A total of four to nine cNFs per patient were treated. An additional two cNFs were included as control tumours. Data were collected in case report forms on the Research Electronic Data Capture (RedCap) platform.
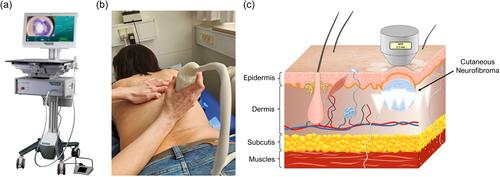
Each selected cNF received one treatment with the HIFU system (System ONE-M, TOOsonix A/S, Hoersholm, Denmark) (Figure 1). A handpiece with a nominal focal depth of 2.3 mm was used. The handpiece was positioned on each cNF to be treated. The exact positioning was displayed as a dermoscopic real-time video feed from a camera housed in the handpiece. The dosing was set to duration 500 ms and acoustic output power of 1.8 W, resulting in a dose energy of 0.9 J/dose. Doses were applied every 1–3 s (video), with approximately 1 mm spacing between each dose to fully cover the tumour with a margin of about 1 mm in the perilesional skin. Local anaesthesia was not used.
Adverse events of the treatment were assessed using six-point grading scales according to the CTCAE system (US Department of Health and Human Services 2017). Patient-reported outcome and patient-reported experience were measured using Likert scales with rating from 0 to 10 or 5-point rating scales of effects (e.g., ranging from absent effect or very satisfied to severe effect or very dissatisfied). To compare the pain in Phase A and Phase B, 2-sample t test was carried out. To estimate the efficacy of treatment, each tumour was evaluated clinically using a 0–10-point scale where 5 was the neutral baseline, 1 major increase in size of the cNF and 10 complete disappearance of the tumour.
The statistical analysis was conducted using The R Foundation for Statistical Computing, Vienna, Austria, http://www.Rproject.org software. Quantitative study variables were described as mean ± SD and qualitative binary variables as n (%). Wilcoxon's rank-sum test was used for two-sample comparisons. Pearson's correlation test was used to analyse relationships between erosions after the treatment and efficacy at 6 months. All tests were two-sided and p < 0.05 was considered as statistically significant.
The primary endpoints were safety and tolerability of the HIFU treatment. The secondary endpoints were other registered biological responses after the treatment, feasibility of the device, change in cNF size and patient's assessment of satisfaction with the treatment and the device.
Seven patients and a total of 68 tumours 2–5 mm in width (median diameter 5 mm) located on the anterior or posterior trunk or on the arms were included in the study. Demographics of the patients and characteristics of the included tumours are shown in Table 1. Of all tumours included, 54 were treated and 14 were untreated controls. The number of doses per lesion varied from 13 to 25; median 18. All enrolled participants completed all required study evaluations.
As observed in the Phase A study, the treatment resulted in mild, local treatment effects.10 Local immediate wheal and flare reactions and oedema were either absent or mild, and no bruising was observed. Experience of pain during the treatment was reported by patients on a 0–10 scale and was graded higher than in Phase A (median 5 vs. 3.5 in Phase A), but without a statistically significant difference (p = 0.400). Pain was instant and disappeared immediately after the dose. At the 6-month follow-up visit, 6/7 participants reported no side effects, and one had mild side effects only. There were no instrument-related adverse events. The primary endpoints concerning safety and tolerability were thus fulfilled.
On follow-up visits, superficial erosions were seen in 24/54 (44%) of tumours during the first week after the treatment. All erosions healed in 1–2 weeks and none developed scars. Pigment changes after 6 months were noted in 10/54 (18.5%) of tumours; all being tumours with erosion after 1 week. Pigment variation with hypopigmentation in the middle surrounded by a hyperpigmented rim was encountered only on skin phototype IV. Summary of observations of pigmentation is included in Table 2. An example of the various biological responses of three cNFs located close to each other in a patient with phototype IV is shown in Figure 2.
The analysis of efficacy 6 months after a single treatment of each cNF as measured using a ruler demonstrated that 70% of the treated cNFs showed at least some reduction in size. Major reduction or complete disappearance was observed in 26% of the tumours (Table 3). The efficacy in Phase B with a median of 6 was lower than the median of 7 in Phase A, but the difference was outside statistical significance (p = 0.09). Further statistical analysis of the data showed significant correlation between reduction in tumour size and observation of a superficial erosion 1 week after treatment (correlation coefficient 0.449, p = 0,005).
This study sought to optimise the dosing of a HIFU treatment which would enable noninvasive therapy of large numbers of cNFs in one session. In Phase A, 250 ms/dose with 0.7 J/dose was used and showed major reduction in 49% of tumours. Although this was encouraging, we tried to further optimise the dosing in Phase B with a dose of 500 ms/dose with 0.9 J/dose. The treatment-related effects remained very mild. The settings used in Phase B did cause a higher level of immediate pain during the treatment. Efficacy as assessed by absolute cNF size tended to be lower in Phase B than Phase A. Interestingly, erosions at 1 week after treatment were strongly correlated with efficacy, indicating that settings in Phase B are preferable in some metrics. Erosions did lead to pigment changes which were most pronounced in patients with darker skin type. The erosions were not seen immediately after treatment but developed within a few days. However, no scarring was encountered.
The limitation of the present study is the small number of patients. That said, 54 cNFs were treated across multiple cNF subtypes and sizes and, therefore, the efficacy seen in this extension supports the tolerability and efficacy seen in the Phase A study. As a result of these data, the 20 MHz HIFU device received CE Mark approval for treatment of cNF in people with NF1 in the European Union. A guidance for use in this context generated from the Phase A and Phase B studies, and a video showing the treatment are provided as supplementary files.
In conclusion, 20 MHz HIFU is demonstrated as a tolerable, safe and efficient method for the treatment of smaller cNFs in patients with NF1. The method is minimally invasive and with potential for clinical use for field eradication of early-stage cNF in different anatomical sites. Given the lack of approved medicinal therapies for cNF as well as the safety and efficacy demonstrated across both the Phases A and B portions of the study, 20 MHz HIFU has been shown to be an important and viable treatment option for early-stage cNFs (Video 1).
Conceptualisation: Sirkku Peltonen, Jørgen Serup, Jaishri O. Blakeley, Katrine Elisabeth Karmisholt and Torsten Bove. Data curation: Sirkku Peltonen, Katrine Elisabeth Karmisholt, Joshua Roberts, Martin Gillstedt and Mimmi Tang. Formal analysis: Sirkku Peltonen, Katrine Elisabeth Karmisholt, Martin Gillstedt, Torsten Bove and Mimmi Tang. Funding acquisition: Sirkku Peltonen, Jørgen Serup, Jaishri O. Blakeley, Torsten Bove and Katrine Elisabeth Karmisholt. Investigation: Sirkku Peltonen, Mimmi Tang and Torsten Bove. Methodology: Sirkku Peltonen, Jørgen Serup, Jaishri O. Blakeley, Torsten Bove, Katrine Elisabeth Karmisholt and Joshua Roberts. Project administration: Sirkku Peltonen, Jørgen Serup, Jaishri O. Blakeley, Joshua Roberts, Torsten Bove and Katrine Elisabeth Karmisholt. Resources: Sirkku Peltonen, Jørgen Serup, Mimmi Tang, Torsten Bove and Katrine Elisabeth Karmisholt. Statistics: Martin Gillstedt and Torsten Bove. Supervision: Sirkku Peltonen, Jørgen Serup, Jaishri O. Blakeley, Joshua Roberts and Katrine Elisabeth Karmisholt. Validation: Sirkku Peltonen, Jørgen Serup, Martin Gillstedt and Katrine Elisabeth Karmisholt. Visualisation: Sirkku Peltonen, Jørgen Serup, Martin Gillstedt, Mimmi Tang, Torsten Bove and Katrine Elisabeth Karmisholt. Writing—Original draft preparation: Sirkku Peltonen, Mimmi Tang and Torsten Bove. Writing—Review, editing and final approval of the manuscript: Sirkku Peltonen, Jørgen Serup, Mimmi Tang, Martin Gillstedt, Jaishri O.Blakeley, Joshua Roberts, Torsten Bove and Katrine Elisabeth Karmisholt.
JS is a sponsor for this clinical investigation funded by NTAP. JOB and JR are officers of NTAP. TB is a shareholder and CEO of TOOsonix A/S. The remaining authors declare no conflict of interest.
The study was approved by the Danish National Ethics Committee, the Swedish Ethics Authority and the Danish and Swedish Medical Agencies. All patients in this manuscript have given written informed consent for participation in the study and the use of their deidentified, anonymized, aggregated data and their case details (including photographs) for publication. The study followed the Good Clinical Practice requirements of ICH.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


