Enantiokonvergente Negishi-Kreuzkupplungen von Racemischen Sekundären Organozink-Reagenzien zur Herstellung Privilegierter Gerüste: Eine Kombinierte Experimentelle und Theoretische Studie
Dr. Alexander Preinfalk, Dr. Rik Oost, Dr. Maximilian F. S. J. Menger, Dr. Marwan Simaan, Dr. Sébastien Lemouzy, Samuel Senoner, Dr. Saad Shaaban, Dr. Boris Maryasin, Prof. Dr. Leticia González, Prof. Dr. Nuno Maulide
{"title":"Enantiokonvergente Negishi-Kreuzkupplungen von Racemischen Sekundären Organozink-Reagenzien zur Herstellung Privilegierter Gerüste: Eine Kombinierte Experimentelle und Theoretische Studie","authors":"Dr. Alexander Preinfalk, Dr. Rik Oost, Dr. Maximilian F. S. J. Menger, Dr. Marwan Simaan, Dr. Sébastien Lemouzy, Samuel Senoner, Dr. Saad Shaaban, Dr. Boris Maryasin, Prof. Dr. Leticia González, Prof. Dr. Nuno Maulide","doi":"10.1002/ange.202414868","DOIUrl":null,"url":null,"abstract":"<p>Enantiokonvergente Reaktionen, bei denen ein racemisches Gemisch in ein enantiomerenangereichertes Produkt umgewandelt wird, stellen eine wichtige Unterklasse im Bereich der asymmetrischen Katalyse dar.<span><sup>1</sup></span> Bis heute existieren mehrere Ansätze, um diese Reaktivität zu erreichen, die im Allgemeinen in zwei mechanistisch unterschiedliche Untergruppen unterteilt werden können: Reaktionen, die eine reversible Racemisierung vor der selektiven Reaktion eines Enantiomers mit dem chiralen Katalysator beinhalten (stereomutative Reaktionen), und Reaktionen, bei denen die chirale Information des Substrats zerstört wird, um ein achirales Zwischenprodukt in situ zu erzeugen. Diese prochirale Spezies interagiert dann mit dem chiralen Katalysator, um ein neues Produkt zu bilden, in sogenannten stereoablativen Prozessen (Schema 1a).<span><sup>1b</sup></span></p><p>Im Kontext enantiokonvergenter Reaktionen stellen Organozink-Verbindungen eine besonders interessante Klasse von Reagenzien dar.<span><sup>1c</sup></span> Während ihre geringere Reaktivität im Vergleich zu anderen organometallischen Nukleophilen wie Organolithium- oder Grignard-Reagenzien eine hohe Funktionalitätsgruppentoleranz gewährleistet, gelten Organozink-Verbindungen oft als konfigurationsstabil aufgrund des hohen Kovalenzgrades der Kohlenstoff-Zink-Bindung.<span><sup>2</sup></span> Diese Tatsache wurde für stereospezifische Kreuzkupplungsreaktionen unter Verwendung konfigurationsdefinierter Organozink-Verbindungen genutzt, wie von Knochel erstmals eingeführt (Schema 1b).<span><sup>3</sup></span> Jedoch racemisieren Organozink-Verbindungen – unter bestimmten Bedingungen – auch in Lösung und können daher für stereokonvergente Prozesse verwendet werden,<span><sup>1c, 4</sup></span> wie von uns in der ersten enantiokonvergenten Fukuyama-Kreuzkupplung eingesetzt.<span><sup>5</sup></span> Diese duale Reaktivität veranlasste uns, unser Verständnis des dynamischen Verhaltens von Organozink-Verbindungen zu erweitern.</p><p>Offene Fragen in diesem Bereich umfassen Folgendes: Welche Elektrophile eignen sich für enantiokonvergente Kreuzkupplungsreaktionen mit Organozink-Nukleophilen, und welche Bedingungen sind erforderlich, um solche Umwandlungen zu erreichen? Welchen Einfluss haben Zusätze auf die stereochemische Labilität von Organozink-Verbindungen? Welcher Mechanismus führt zur Inversion der Konfiguration an der Kohlenstoff-Zink-Bindung?</p><p>Insbesondere konzentrierten wir unsere Aufmerksamkeit auf einfache Aryl-Elektrophile. Dies würde einen effektiven Zugang zu 1,1-Diarylalkenen ermöglichen, einer privilegierten Struktur in Arzneimitteln (Schema 1c).<span><sup>6, 7</sup></span> Hiermit präsentieren wir einen enantiokonvergenten Negishi-Kreuzkupplungsansatz (Schema 1d), um auf dieses Gerüst zuzugreifen, basierend auf einem einzigartigen stereomutativen Mechanismus der dynamischen kinetischen Racematspaltung (DKR).</p><p>Wir begannen unsere Untersuchung mit den zuvor entwickelten Bedingungen unter Verwendung des benzylichen Organozink-Reagenz <b>2 a</b>, Palladiumacetat als Palladiumquelle und einem leichten Überschuss an Ligand in Methyl-<i>tert</i>-butylether (MTBE) als Lösungsmittel bei Raumtemperatur (Schema 2). Basierend auf einem zentralen Befund von Knochel, dass das Hinzufügen von Zinkhalogeniden zu einer Erosion der Enantioselektivität bei stereospezifischen Kreuzkupplungsreaktionen führt,<span><sup>8</sup></span> stellten wir die Hypothese auf, dass diese schnellere Inversion der Kohlenstoff-Zink-Bindung für unser System vorteilhaft sein sollte. Daher verwendeten wir Zinkchlorid als Zusatzstoff. Unter Verwendung von Ethyl-4-iodbenzoat <b>1 a</b> als Modell-Elektrophil und dem TADDOL-basierten Phosphoramidit-Liganden <b>L1</b>, der sich in der Fukuyama-Kreuzkupplung am besten bewährte, wurde das gewünschte 1,1-Diarylethan <b>3 a</b> in hoher Ausbeute gebildet. Zu unserer Überraschung war das Produkt fast racemisch. Daraufhin entschieden wir uns, chirale Amin-Rückgrate in das Ligandensystem einzuführen, was jedoch zu keiner Verbesserung führte (<b>L2</b> und <b>L3</b>). Im weiteren Verlauf untersuchten wir Phosphit-Liganden. Während die anfänglichen Ergebnisse mit diesem Ligandentyp, der entweder einen chiralen Alkohol (<b>L4</b>) oder ein chirales Diol (<b>L5</b>) trägt, enttäuschend waren, stellten wir fest, dass eine Kombination aus zwei chiralen Bausteinen vielversprechender war (<b>L6</b>–<b>L9</b>).</p><p>Da bekannt ist, dass Ate-Komplexe schnellere Transmetallierungsraten aufweisen,<span><sup>9</sup></span> stellten wir die Hypothese auf, dass eine höhere Enantioselektivität erreicht werden könnte, wenn das Organozink-Reagenz in Abwesenheit von Lithiumsalzen hergestellt wird. Tatsächlich führten Reaktionen mit dem lithiumfreien Zink-Reagenz <b>2 a’</b> zu höheren Enantiomerenüberschüssen (Tabelle 1, Eintrag 1). Anschließend untersuchten wir den Einfluss von Substituenten auf das Phosphit-Rückgrat. Die Änderung der Arylgruppe im TADDOL-Motiv führte zu einer Verringerung des Enantiomerenverhältnisses <i>e. r</i>. (<b>L10</b>, Eintrag 2), was darauf hinweist, dass die <i>p-tert-</i>Butylgruppe, die wir seit Beginn unserer Studie verwendet haben, einen vorteilhaften Einfluss auf das Reaktionsergebnis hat. Die Änderung des Mentholgerüsts zu kommerziell erhältlichem Phenylmenthol führte zu einer Erhöhung des <i>e. r</i>. (<b>L11</b>, Eintrag 3).\n</p><p>Anschließend untersuchten wir weitere Parameter, einschließlich des Lösungsmittels, der Abgangsgruppe am Elektrophil, der Palladiumquelle und des Palladium/Ligand-Verhältnisses. MTBE erwies sich als das beste Lösungsmittel (siehe Hintergrundinformationen für eine detaillierte Studie), Palladiumcinnamylchlorid als die kompetenteste Palladiumquelle, und ein 3 : 1-Verhältnis von Ligand zu Palladium ist ideal. Wir stellten außerdem fest, dass Chloride unreaktiv sind (Eintrag 4), während das Verlangsamen der oxidativen Addition durch die Verwendung eines Aryl-Bromids einen positiven Effekt auf den <i>e. r</i>.-Wert hatte (Eintrag 5).</p><p>Die Verwendung sperrigerer Derivate von Phenylmenthol am Liganden führte überraschenderweise zu einer leichten Verringerung des <i>e. r</i>. (Eintrag 6). Die Änderung der Art des Nukleophils zum entsprechenden Organozinkbromid führte zu vergleichbaren Enantioselektivitätswerten, jedoch zu einer geringeren Ausbeute (Eintrag 7). Zuletzt führten wir die Reaktion bei verschiedenen Temperaturen durch (Einträge 8 und 9) und stellten fest, dass eine Reaktionstemperatur von 21 °C ideal ist (Eintrag 9).</p><p>Anschließend untersuchten wir das Reaktionsspektrum (Schema 3). Am Elektrophil wurden mehrere Substituenten in <i>ortho-</i> und <i>meta</i>-Position gut toleriert. In <i>para</i>-Position führten elektronenarme Substituenten (<b>3 a</b>–<b>3 c</b>) sowie Alkine (<b>3 d</b>), Alkene (<b>3 e</b>) und Arylgruppen (<b>3 f</b>) zu den gewünschten Produkten in hervorragender Ausbeute und sehr guten <i>e. r.-</i>Werten. Thioether- und Ethergruppen (<b>3 g</b> und <b>3 j</b>) führten zu den gewünschten Produkten mit vergleichbaren Enantioselektivitätswerten. In <i>meta-</i>Position wurden ähnliche Ergebnisse mit Thioether- und Chloridsubstituenten (<b>3 h</b> und <b>3 i</b>) erzielt. Disubstituierte Aromaten, einschließlich <i>meta-para-</i> und <i>meta-meta</i>-Substituenten, wurden gut toleriert (<b>3 k</b>–<b>3 m</b>), darunter auch potenziell reaktive cyclische Ketone (<b>3 l</b>). Bemerkenswerterweise erwiesen sich benzylische Chloride als geeignete Substrate (<b>3 n</b>) und lieferten Produkte, die mit zuvor entwickelten nickelbasierten Ansätzen nicht zugänglich sind.<span><sup>7a, 7d</sup></span></p><p>Wir untersuchten als Nächstes das Substratspektrum der Organozink-Nukleophile. Bei der Alkylkomponente führten sperrigere Systeme zu einem leichten Anstieg des Enantiomerenüberschusses (<b>3 o</b>–<b>3 q</b>), wobei das gewünschte Produkt in einem Enantiomerenverhältnis von bis zu 90 : 10 erhalten wurde (<b>3 p</b>). Schließlich führte die Kombination eines fluorhaltigen Organozink-Nukleophils mit mehreren fluorhaltigen Elektrophilen zur Bildung einer potenziell interessanten Bibliothek von Diaryl-Motiven, wobei hervorragende Ausbeuten und Enantiomerenverhältnisse der gewünschten Produkte erzielt wurden (<b>3 r</b>–<b>3 y</b>). Bemerkenswerterweise führte benzylisches Bromid mit <i>ortho</i>-Substitution zu einer geringeren Ausbeute und einem niedrigeren ee-Wert (<b>3 z</b>).</p><p>Zusammenfassend haben wir ein Verfahren für die enantiokonvergente Negishi-Kreuzkupplung von racemischen sekundären Organozink-Reagenzien mit Aryl-Elektrophilen unter Verwendung neuer Ligandengerüste entwickelt. Nach weiterer Optimierung der Parameter konnte die Allgemeingültigkeit unseres neu entwickelten Verfahrens anhand eines breiten Substratspektrums demonstriert werden, wobei die entsprechenden 1,1-Diarylalkane in der Regel in hoher Ausbeute und mit moderaten bis guten Enantioselektivitäten erhalten wurden.</p><p>Wir haben darüber hinaus unser Verständnis dieser Reaktion durch computergestützte Studien erweitert. Dabei stellten wir fest, dass der Hauptgrund für die geringere Enantioselektivität nicht, wie erwartet, eine schwache enantiomere Induktion durch den Liganden ist, sondern vielmehr eine konkurrierende Ligandendissoziation, die zum racemischen Produkt führt. Unsere Berechnungen deuten zudem darauf hin, dass die Racemisierung von Organozink-Verbindungen höchstwahrscheinlich über einen gebrückten bimolekularen Mechanismus erfolgt, was den Einfluss von Zinkhalogeniden auf die Konfigurationsstabilität von Organozink-Verbindungen erklärt.</p><p>Die Autoren haben weitere Referenzen in den Hintergrundinformationen zitiert.<span><sup>14-22, 23-48</sup></span></p><p>Die Autoren erklären, dass keine Interessenkonflikte vorliegen.</p>","PeriodicalId":7803,"journal":{"name":"Angewandte Chemie","volume":"136 50","pages":""},"PeriodicalIF":0.0000,"publicationDate":"2024-11-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/ange.202414868","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Angewandte Chemie","FirstCategoryId":"1085","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/ange.202414868","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
Enantiokonvergente Reaktionen, bei denen ein racemisches Gemisch in ein enantiomerenangereichertes Produkt umgewandelt wird, stellen eine wichtige Unterklasse im Bereich der asymmetrischen Katalyse dar.1 Bis heute existieren mehrere Ansätze, um diese Reaktivität zu erreichen, die im Allgemeinen in zwei mechanistisch unterschiedliche Untergruppen unterteilt werden können: Reaktionen, die eine reversible Racemisierung vor der selektiven Reaktion eines Enantiomers mit dem chiralen Katalysator beinhalten (stereomutative Reaktionen), und Reaktionen, bei denen die chirale Information des Substrats zerstört wird, um ein achirales Zwischenprodukt in situ zu erzeugen. Diese prochirale Spezies interagiert dann mit dem chiralen Katalysator, um ein neues Produkt zu bilden, in sogenannten stereoablativen Prozessen (Schema 1a).1b
Im Kontext enantiokonvergenter Reaktionen stellen Organozink-Verbindungen eine besonders interessante Klasse von Reagenzien dar.1c Während ihre geringere Reaktivität im Vergleich zu anderen organometallischen Nukleophilen wie Organolithium- oder Grignard-Reagenzien eine hohe Funktionalitätsgruppentoleranz gewährleistet, gelten Organozink-Verbindungen oft als konfigurationsstabil aufgrund des hohen Kovalenzgrades der Kohlenstoff-Zink-Bindung.2 Diese Tatsache wurde für stereospezifische Kreuzkupplungsreaktionen unter Verwendung konfigurationsdefinierter Organozink-Verbindungen genutzt, wie von Knochel erstmals eingeführt (Schema 1b).3 Jedoch racemisieren Organozink-Verbindungen – unter bestimmten Bedingungen – auch in Lösung und können daher für stereokonvergente Prozesse verwendet werden,1c, 4 wie von uns in der ersten enantiokonvergenten Fukuyama-Kreuzkupplung eingesetzt.5 Diese duale Reaktivität veranlasste uns, unser Verständnis des dynamischen Verhaltens von Organozink-Verbindungen zu erweitern.
Offene Fragen in diesem Bereich umfassen Folgendes: Welche Elektrophile eignen sich für enantiokonvergente Kreuzkupplungsreaktionen mit Organozink-Nukleophilen, und welche Bedingungen sind erforderlich, um solche Umwandlungen zu erreichen? Welchen Einfluss haben Zusätze auf die stereochemische Labilität von Organozink-Verbindungen? Welcher Mechanismus führt zur Inversion der Konfiguration an der Kohlenstoff-Zink-Bindung?
Insbesondere konzentrierten wir unsere Aufmerksamkeit auf einfache Aryl-Elektrophile. Dies würde einen effektiven Zugang zu 1,1-Diarylalkenen ermöglichen, einer privilegierten Struktur in Arzneimitteln (Schema 1c).6, 7 Hiermit präsentieren wir einen enantiokonvergenten Negishi-Kreuzkupplungsansatz (Schema 1d), um auf dieses Gerüst zuzugreifen, basierend auf einem einzigartigen stereomutativen Mechanismus der dynamischen kinetischen Racematspaltung (DKR).
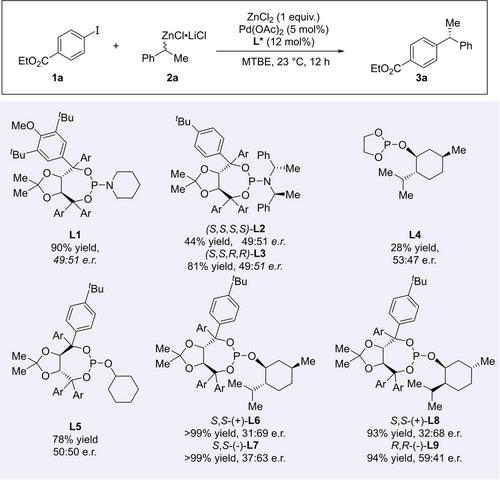
Wir begannen unsere Untersuchung mit den zuvor entwickelten Bedingungen unter Verwendung des benzylichen Organozink-Reagenz 2 a, Palladiumacetat als Palladiumquelle und einem leichten Überschuss an Ligand in Methyl-tert-butylether (MTBE) als Lösungsmittel bei Raumtemperatur (Schema 2). Basierend auf einem zentralen Befund von Knochel, dass das Hinzufügen von Zinkhalogeniden zu einer Erosion der Enantioselektivität bei stereospezifischen Kreuzkupplungsreaktionen führt,8 stellten wir die Hypothese auf, dass diese schnellere Inversion der Kohlenstoff-Zink-Bindung für unser System vorteilhaft sein sollte. Daher verwendeten wir Zinkchlorid als Zusatzstoff. Unter Verwendung von Ethyl-4-iodbenzoat 1 a als Modell-Elektrophil und dem TADDOL-basierten Phosphoramidit-Liganden L1, der sich in der Fukuyama-Kreuzkupplung am besten bewährte, wurde das gewünschte 1,1-Diarylethan 3 a in hoher Ausbeute gebildet. Zu unserer Überraschung war das Produkt fast racemisch. Daraufhin entschieden wir uns, chirale Amin-Rückgrate in das Ligandensystem einzuführen, was jedoch zu keiner Verbesserung führte (L2 und L3). Im weiteren Verlauf untersuchten wir Phosphit-Liganden. Während die anfänglichen Ergebnisse mit diesem Ligandentyp, der entweder einen chiralen Alkohol (L4) oder ein chirales Diol (L5) trägt, enttäuschend waren, stellten wir fest, dass eine Kombination aus zwei chiralen Bausteinen vielversprechender war (L6–L9).
Da bekannt ist, dass Ate-Komplexe schnellere Transmetallierungsraten aufweisen,9 stellten wir die Hypothese auf, dass eine höhere Enantioselektivität erreicht werden könnte, wenn das Organozink-Reagenz in Abwesenheit von Lithiumsalzen hergestellt wird. Tatsächlich führten Reaktionen mit dem lithiumfreien Zink-Reagenz 2 a’ zu höheren Enantiomerenüberschüssen (Tabelle 1, Eintrag 1). Anschließend untersuchten wir den Einfluss von Substituenten auf das Phosphit-Rückgrat. Die Änderung der Arylgruppe im TADDOL-Motiv führte zu einer Verringerung des Enantiomerenverhältnisses e. r. (L10, Eintrag 2), was darauf hinweist, dass die p-tert-Butylgruppe, die wir seit Beginn unserer Studie verwendet haben, einen vorteilhaften Einfluss auf das Reaktionsergebnis hat. Die Änderung des Mentholgerüsts zu kommerziell erhältlichem Phenylmenthol führte zu einer Erhöhung des e. r. (L11, Eintrag 3).
Anschließend untersuchten wir weitere Parameter, einschließlich des Lösungsmittels, der Abgangsgruppe am Elektrophil, der Palladiumquelle und des Palladium/Ligand-Verhältnisses. MTBE erwies sich als das beste Lösungsmittel (siehe Hintergrundinformationen für eine detaillierte Studie), Palladiumcinnamylchlorid als die kompetenteste Palladiumquelle, und ein 3 : 1-Verhältnis von Ligand zu Palladium ist ideal. Wir stellten außerdem fest, dass Chloride unreaktiv sind (Eintrag 4), während das Verlangsamen der oxidativen Addition durch die Verwendung eines Aryl-Bromids einen positiven Effekt auf den e. r.-Wert hatte (Eintrag 5).
Die Verwendung sperrigerer Derivate von Phenylmenthol am Liganden führte überraschenderweise zu einer leichten Verringerung des e. r. (Eintrag 6). Die Änderung der Art des Nukleophils zum entsprechenden Organozinkbromid führte zu vergleichbaren Enantioselektivitätswerten, jedoch zu einer geringeren Ausbeute (Eintrag 7). Zuletzt führten wir die Reaktion bei verschiedenen Temperaturen durch (Einträge 8 und 9) und stellten fest, dass eine Reaktionstemperatur von 21 °C ideal ist (Eintrag 9).
Anschließend untersuchten wir das Reaktionsspektrum (Schema 3). Am Elektrophil wurden mehrere Substituenten in ortho- und meta-Position gut toleriert. In para-Position führten elektronenarme Substituenten (3 a–3 c) sowie Alkine (3 d), Alkene (3 e) und Arylgruppen (3 f) zu den gewünschten Produkten in hervorragender Ausbeute und sehr guten e. r.-Werten. Thioether- und Ethergruppen (3 g und 3 j) führten zu den gewünschten Produkten mit vergleichbaren Enantioselektivitätswerten. In meta-Position wurden ähnliche Ergebnisse mit Thioether- und Chloridsubstituenten (3 h und 3 i) erzielt. Disubstituierte Aromaten, einschließlich meta-para- und meta-meta-Substituenten, wurden gut toleriert (3 k–3 m), darunter auch potenziell reaktive cyclische Ketone (3 l). Bemerkenswerterweise erwiesen sich benzylische Chloride als geeignete Substrate (3 n) und lieferten Produkte, die mit zuvor entwickelten nickelbasierten Ansätzen nicht zugänglich sind.7a, 7d
Wir untersuchten als Nächstes das Substratspektrum der Organozink-Nukleophile. Bei der Alkylkomponente führten sperrigere Systeme zu einem leichten Anstieg des Enantiomerenüberschusses (3 o–3 q), wobei das gewünschte Produkt in einem Enantiomerenverhältnis von bis zu 90 : 10 erhalten wurde (3 p). Schließlich führte die Kombination eines fluorhaltigen Organozink-Nukleophils mit mehreren fluorhaltigen Elektrophilen zur Bildung einer potenziell interessanten Bibliothek von Diaryl-Motiven, wobei hervorragende Ausbeuten und Enantiomerenverhältnisse der gewünschten Produkte erzielt wurden (3 r–3 y). Bemerkenswerterweise führte benzylisches Bromid mit ortho-Substitution zu einer geringeren Ausbeute und einem niedrigeren ee-Wert (3 z).
Zusammenfassend haben wir ein Verfahren für die enantiokonvergente Negishi-Kreuzkupplung von racemischen sekundären Organozink-Reagenzien mit Aryl-Elektrophilen unter Verwendung neuer Ligandengerüste entwickelt. Nach weiterer Optimierung der Parameter konnte die Allgemeingültigkeit unseres neu entwickelten Verfahrens anhand eines breiten Substratspektrums demonstriert werden, wobei die entsprechenden 1,1-Diarylalkane in der Regel in hoher Ausbeute und mit moderaten bis guten Enantioselektivitäten erhalten wurden.
Wir haben darüber hinaus unser Verständnis dieser Reaktion durch computergestützte Studien erweitert. Dabei stellten wir fest, dass der Hauptgrund für die geringere Enantioselektivität nicht, wie erwartet, eine schwache enantiomere Induktion durch den Liganden ist, sondern vielmehr eine konkurrierende Ligandendissoziation, die zum racemischen Produkt führt. Unsere Berechnungen deuten zudem darauf hin, dass die Racemisierung von Organozink-Verbindungen höchstwahrscheinlich über einen gebrückten bimolekularen Mechanismus erfolgt, was den Einfluss von Zinkhalogeniden auf die Konfigurationsstabilität von Organozink-Verbindungen erklärt.
Die Autoren haben weitere Referenzen in den Hintergrundinformationen zitiert.14-22, 23-48
Die Autoren erklären, dass keine Interessenkonflikte vorliegen.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


