Zhu-Zhu Sun, Yang Li, Yunhai Zhang, Quan-Song Li and Wei-Lu Ding
{"title":"Thiadiazolophenanthroline-based hole-transporter for durable and efficient perovskite solar cells: atomic-level insights for performance enhancement†","authors":"Zhu-Zhu Sun, Yang Li, Yunhai Zhang, Quan-Song Li and Wei-Lu Ding","doi":"10.1039/D4NJ04021J","DOIUrl":null,"url":null,"abstract":"<p >Finding remarkable hole-transporting materials (HTMs) for perovskite solar cells (PSCs) is crucial but challenging, and rationally regulating the acceptor structure is one of the most effective strategies. In this work, a novel electron-withdrawing moiety of thiadiazolophenanthroline (TPT) was first exploited as the acceptor structure of donor–acceptor–donor (D–A–D)-type HTMs. The isolated molecular and interfacial properties of TPT-based HTMs (SM-4) were methodically investigated by comparing with the simulated results of benzothiadiazole (BT, SM-1) and phenanthrothiadiazole (PT, SM-2) acceptors. Theoretical simulations manifest that SM-4 displays a more negative HOMO energy and larger hole mobility than SM-1 and SM-2. The higher mobility of SM-4 is derived from the larger hole transfer integral due to easier intermolecular orbital overlap. Moreover, the better optical excitation property, smaller exciton binding energy, and profitable solubility and stability are shown for SM-4. Furthermore, interfacial calculations reveal that advantageous photon-induced excitation dissociation can be anticipated at the interface because of greater interfacial charge redistribution and more suitable energy levels. Overall, our simulations suggest that the designed TPT-based acceptor molecule holds great promise as a potential HTM candidate, providing support for more efficient PSCs.</p>","PeriodicalId":95,"journal":{"name":"New Journal of Chemistry","volume":" 47","pages":" 20051-20060"},"PeriodicalIF":2.5000,"publicationDate":"2024-11-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"New Journal of Chemistry","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/nj/d4nj04021j","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
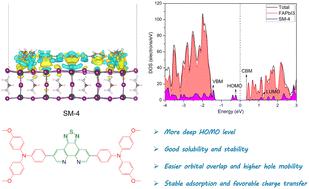
Finding remarkable hole-transporting materials (HTMs) for perovskite solar cells (PSCs) is crucial but challenging, and rationally regulating the acceptor structure is one of the most effective strategies. In this work, a novel electron-withdrawing moiety of thiadiazolophenanthroline (TPT) was first exploited as the acceptor structure of donor–acceptor–donor (D–A–D)-type HTMs. The isolated molecular and interfacial properties of TPT-based HTMs (SM-4) were methodically investigated by comparing with the simulated results of benzothiadiazole (BT, SM-1) and phenanthrothiadiazole (PT, SM-2) acceptors. Theoretical simulations manifest that SM-4 displays a more negative HOMO energy and larger hole mobility than SM-1 and SM-2. The higher mobility of SM-4 is derived from the larger hole transfer integral due to easier intermolecular orbital overlap. Moreover, the better optical excitation property, smaller exciton binding energy, and profitable solubility and stability are shown for SM-4. Furthermore, interfacial calculations reveal that advantageous photon-induced excitation dissociation can be anticipated at the interface because of greater interfacial charge redistribution and more suitable energy levels. Overall, our simulations suggest that the designed TPT-based acceptor molecule holds great promise as a potential HTM candidate, providing support for more efficient PSCs.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


