Emerging concepts and treatments in autoinflammatory interferonopathies and monogenic systemic lupus erythematosus
IF 29.4
1区 医学
Q1 RHEUMATOLOGY
引用次数: 0
Abstract
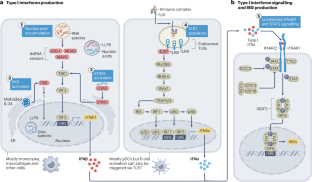
Over the past two decades, the number of genetically defined autoinflammatory interferonopathies has steadily increased. Aicardi–Goutières syndrome and proteasome-associated autoinflammatory syndromes (PRAAS, also known as CANDLE) are caused by genetic defects that impair homeostatic intracellular nucleic acid and protein processing respectively. Research into these genetic defects revealed intracellular sensors that activate type I interferon production. In SAVI and COPA syndrome, genetic defects that cause chronic activation of the dinucleotide sensor stimulator of interferon genes (STING) share features of lung inflammation and fibrosis; and selected mutations that amplify interferon-α/β receptor signalling cause central nervous system manifestations resembling Aicardi–Goutières syndrome. Research into the monogenic causes of childhood-onset systemic lupus erythematosus (SLE) demonstrates the pathogenic role of autoantibodies to particle-bound extracellular nucleic acids that distinguishes monogenic SLE from the autoinflammatory interferonopathies. This Review introduces a classification for autoinflammatory interferonopathies and discusses the divergent and shared pathomechanisms of interferon production and signalling in these diseases. Early success with drugs that block type I interferon signalling, new insights into the roles of cytoplasmic DNA or RNA sensors, pathways in type I interferon production and organ-specific pathology of the autoinflammatory interferonopathies and monogenic SLE, reveal novel drug targets that could personalize treatment approaches. This Review provides a comprehensive update on dysregulated type I interferon production and signalling in autoinflammatory interferonopathies, monogenic systemic lupus erythematosus and conditions that present with broad immune dysregulation and interferon signatures. The authors provide a classification for autoinflammatory interferonopathies based on disease mechanisms of increased type I interferon production and signalling and overlapping clinical phenotypes.

自体炎性干扰素病变和单基因系统性红斑狼疮的新概念和治疗
在过去的二十年中,基因定义的自身炎症性干扰素病变的数量稳步增加。aicardii - gouti综合征和蛋白酶体相关自身炎症综合征(PRAAS,也称为CANDLE)是由基因缺陷引起的,它们分别损害细胞内稳态核酸和蛋白质加工。对这些遗传缺陷的研究揭示了激活I型干扰素产生的细胞内传感器。在SAVI和COPA综合征中,导致干扰素基因二核苷酸传感器刺激因子(STING)慢性激活的遗传缺陷具有肺部炎症和纤维化的特征;以及放大干扰素-α/β受体信号传导的特定突变,导致类似aicardii - gouti综合征的中枢神经系统表现。对儿童期系统性红斑狼疮(SLE)单基因病因的研究表明,颗粒结合细胞外核酸自身抗体的致病作用将单基因SLE与自身炎症性干扰素病区分开来。本文介绍了自身炎症性干扰素病变的分类,并讨论了这些疾病中干扰素产生和信号传导的不同和共同的病理机制。阻断I型干扰素信号传导的药物的早期成功,细胞质DNA或RNA传感器作用的新见解,I型干扰素产生的途径以及自身炎症性干扰素病变和单基因SLE的器官特异性病理,揭示了新的药物靶点,可以个性化治疗方法。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Nature Reviews Rheumatology
医学-风湿病学
CiteScore
29.90
自引率
0.90%
发文量
137
审稿时长
6-12 weeks
期刊介绍:
Nature Reviews Rheumatology is part of the Nature Reviews portfolio of journals. The journal scope covers the entire spectrum of rheumatology research. We ensure that our articles are accessible to the widest possible audience.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


