DFT Studies on the Mechanism of Ligand-Regulated Palladium-Catalyzed Iodide-Assisted Hydrocarbonylation of Olefins with Formic Acid: Favored Reaction Routes and Selectivities
IF 3.6
2区 化学
Q1 CHEMISTRY, ORGANIC
引用次数: 0
Abstract
DFT calculations have been performed to gain insight into the mechanism of hydrocarbonylation of olefins and the origin of regio- and chemoselectivity. It is shown that the most feasible mechanism involves five steps: (i) decomposition of acetic formic anhydride, (ii) hydropalladation of olefins, (iii) CO migratory insertion, (iv) iodide-assisted acetate-formate exchange, and (v) formylation or carboxylation. Importantly, carboxylation proceeds via the decomposition of anhydride, followed by reductive elimination instead of direct hydrolysis of anhydride. For phosphine-ligated palladium catalysis, on one hand, the lower stability of the transition state leading to 1,2-hydropalladation could be attributed to H···H steric hindrance. On the other hand, the high chemoselectivity for the aldehyde is ascribed to increased π back-donation effect and ligand-substrate noncovalent interactions, which stabilize the transition state and hence reduce the energy barrier. For ferrocenyl phosphine-ligated palladium catalysis, significant C–H···π interaction between the substrate and proximal phenyl moiety of the phenylphosphine and π–π interaction between formate and phenyl moiety can facilitate the carboxylation process. This in-depth mechanistic insight can account for reactivity and selectivity at an atomistic level and have implications for designing new generations of palladium catalysts.
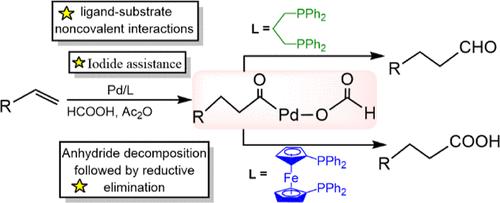
配体调控钯催化碘辅助烯烃与甲酸羰基化反应机理的DFT研究:有利的反应途径和选择性
进行了DFT计算,以深入了解烯烃烃基化的机理和区域选择性和化学选择性的起源。结果表明,最可行的机理包括五个步骤:(i)乙酸甲酸酐的分解,(ii)烯烃的氢钯化,(iii) CO迁移插入,(iv)碘化物辅助醋酸酯-甲酸交换,(v)甲酰化或羧基化。重要的是,羧基化是通过酸酐的分解进行的,然后是还原消除,而不是直接水解酸酐。对于膦连接钯的催化,一方面,导致1,2-氢钯化的过渡态稳定性较低可归因于H···H位阻。另一方面,醛的高化学选择性归因于π反给能效应和配体-底物非共价相互作用的增加,这些相互作用稳定了过渡态,从而降低了能垒。在二茂铁膦连接钯催化反应中,底物与苯基膦近端苯基段之间存在显著的C-H··π相互作用,甲酸酯与苯基段之间存在显著的π -π相互作用,有利于羧基化反应的进行。这种深入的机理见解可以在原子水平上解释反应性和选择性,并对设计新一代钯催化剂具有指导意义。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Journal of Organic Chemistry
化学-有机化学
CiteScore
6.20
自引率
11.10%
发文量
1467
审稿时长
2 months
期刊介绍:
Journal of Organic Chemistry welcomes original contributions of fundamental research in all branches of the theory and practice of organic chemistry. In selecting manuscripts for publication, the editors place emphasis on the quality and novelty of the work, as well as the breadth of interest to the organic chemistry community.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


