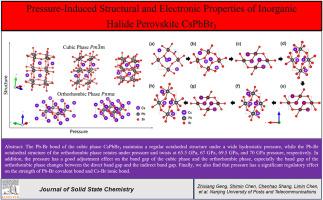
Pressure-induced structural and electronic properties of inorganic halide perovskite CsPbBr3
IF 3.2
3区 化学
Q2 CHEMISTRY, INORGANIC & NUCLEAR
引用次数: 0
Abstract
In this paper, Density Functional Theory (DFT) based on first principles is used to study the influence of pressure on the structural and electronic properties of the cubic and orthorhombic phases of halogen perovskite CsPbBr3. We observe that the lattice constant of the cubic phase shows a monotonically decreasing trend in a wide pressure range, while the Pb–Br irregular octahedron of the orthorhombic phase rotates under pressure and distorts at 65.5 GPa, 67 GPa, 69.5 GPa, and 70 GPa pressure points, respectively. Moreover, the pressure has a good adjustment effect on the band gap of the cubic and orthorhombic phases. The band gap of the orthorhombic phase changes between the direct band gap and the indirect band gap under pressure. The charge difference density and density of states are calculated to study the type of bonds between internal atoms and the effect of pressure on the distribution of orbital electrons. The study of the Crystal Orbital Hamilton Populations and lattice energy of the electron cloud shows that the pressure has a significant adjustment effect on the strength of the Pb–Br covalent bond and the Cs–Br ionic bond. These results provide a comprehensive reference and guidance for the further research and application of perovskite material pressure engineering.
无机卤化物钙钛矿CsPbBr3的压力诱导结构和电子性能
本文采用基于第一性原理的密度泛函理论(DFT)研究了压力对卤素钙钛矿CsPbBr3立方相和正交相结构和电子性能的影响。我们观察到立方相的晶格常数在较宽的压力范围内呈单调减小的趋势,而正交相的Pb-Br不规则八面体在压力下旋转,并分别在65.5 GPa、67 GPa、69.5 GPa和70 GPa压力点处扭曲。此外,压力对立方相和正交相的带隙有很好的调节作用。在压力作用下,正交相的带隙在直接带隙和间接带隙之间变化。计算了电荷差密度和态密度,研究了内部原子间键的类型和压力对轨道电子分布的影响。晶体轨道汉密尔顿居群和电子云晶格能的研究表明,压力对Pb-Br共价键和Cs-Br离子键的强度有显著的调节作用。这些结果为钙钛矿材料压力工程的进一步研究和应用提供了全面的参考和指导。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Journal of Solid State Chemistry
化学-无机化学与核化学
CiteScore
6.00
自引率
9.10%
发文量
848
审稿时长
25 days
期刊介绍:
Covering major developments in the field of solid state chemistry and related areas such as ceramics and amorphous materials, the Journal of Solid State Chemistry features studies of chemical, structural, thermodynamic, electronic, magnetic, and optical properties and processes in solids.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


