Identification of inhibitors targeting the FLT3-ITD mutation through 4D-QSAR, in vitro, and in silico
IF 6
2区 医学
Q1 CHEMISTRY, MEDICINAL
引用次数: 0
Abstract
The FMS-like tyrosine kinase 3-internal tandem duplication (FLT3-ITD) mutation is a key target for acute myeloid leukemia (AML) treatment. The second-generation inhibitors such as Gilteritinib still present off-target effects and associated side effects. Therefore, identifying novel FLT3-ITD inhibitors has become a promising strategy for AML treatment. In this study, a 4D-QSAR model was developed based on Gilteritinib and its analogues, and it was found that introducing hydrophobic bulky groups at the piperazine or piperidine of Gilteritinib would enhance the binding affinity to FLT3-ITD. So, three series of targeted compounds (A1-A5, B1–B5 and C1–C5) were designed and synthesized. The antiproliferative activity against MOLM-13 cells was evaluated in vitro. Compound A1 (IC50 = 25.65 nM), with a cubane group at the piperazine position; Compounds B2 (IC50 = 63.38 nM) and C2 (IC50 = 54.96 nM), with a norbornene group at the piperidine position, showed the strongest inhibition in their series. Their IC50 values were comparable to that of the positive control Gilteritinib (IC50 = 22.37 nM). FLT3-ITD was confirmed as the degradation target through a kinase inhibition assay, where the IC50 values were 2.12 nM (Compound A1), 1.29 nM (Compound B2), and 3.06 nM (Compound C2), which were comparable to that of Gilteritinib (IC50 = 0.43 nM). Additionally, molecular docking and molecular dynamics (MD) simulations showed that Compounds A1, B2, and C2 had similar binding modes to that of Gilteritinib with more stable affinities. Overall, these results demonstrated that Compounds A1, B2, and C2 were promising inhibitors for targeting AML with FLT3-ITD mutation.
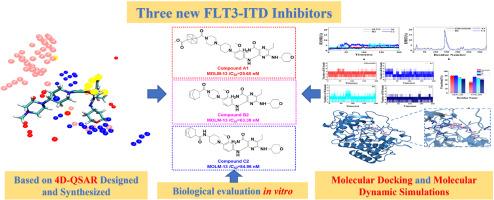
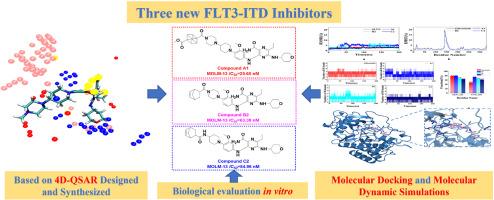
通过4D-QSAR、体外和硅学鉴定针对FLT3-ITD突变的抑制剂
FMS样酪氨酸激酶3-内部串联重复(FLT3-ITD)突变是急性髓性白血病(AML)治疗的关键靶点。吉利替尼等第二代抑制剂仍存在脱靶效应和相关副作用。因此,寻找新型的FLT3-ITD抑制剂已成为治疗急性髓性白血病的有效策略。本研究以吉瑞替尼及其类似物为基础,建立了4D-QSAR模型,发现在吉瑞替尼的哌嗪或哌啶上引入疏水性大基团可增强与FLT3-ITD的结合亲和力。因此,设计并合成了三个系列的靶向化合物(A1-A5、B1-B5 和 C1-C5)。对 MOLM-13 细胞的抗增殖活性进行了体外评估。化合物 A1(IC50 = 25.65 nM)在哌嗪位置上带有一个立方烷基团;化合物 B2(IC50 = 63.38 nM)和 C2(IC50 = 54.96 nM)在哌啶位置上带有一个降冰片烯基团。它们的 IC50 值与阳性对照 Gilteritinib(IC50 = 22.37 nM)相当。通过激酶抑制实验确认了 FLT3-ITD 为降解靶点,其 IC50 值分别为 2.12 nM(化合物 A1)、1.29 nM(化合物 B2)和 3.06 nM(化合物 C2),与吉特替尼(IC50 = 0.43 nM)相当。此外,分子对接和分子动力学(MD)模拟显示,化合物 A1、B2 和 C2 与吉特替尼的结合模式相似,亲和力更稳定。总之,这些结果表明化合物 A1、B2 和 C2 是针对 FLT3-ITD 突变的急性髓细胞性白血病很有前景的抑制剂。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

CiteScore
11.70
自引率
9.00%
发文量
863
审稿时长
29 days
期刊介绍:
The European Journal of Medicinal Chemistry is a global journal that publishes studies on all aspects of medicinal chemistry. It provides a medium for publication of original papers and also welcomes critical review papers.
A typical paper would report on the organic synthesis, characterization and pharmacological evaluation of compounds. Other topics of interest are drug design, QSAR, molecular modeling, drug-receptor interactions, molecular aspects of drug metabolism, prodrug synthesis and drug targeting. The journal expects manuscripts to present the rational for a study, provide insight into the design of compounds or understanding of mechanism, or clarify the targets.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


