A. Waleed, D. Younus, A. A. Issa, H. R. Obayes, M. S. Ali, D. S. El-Sayed
{"title":"Adsorption Locator Behavior of Polycyclic-Carbon Based Systems: Computational Optical and Dynamic Properties","authors":"A. Waleed, D. Younus, A. A. Issa, H. R. Obayes, M. S. Ali, D. S. El-Sayed","doi":"10.1134/S107036322410013X","DOIUrl":null,"url":null,"abstract":"<p>Polycyclic systems such as acenaphthylene, fluoranthene, benzo[<i>k</i>]fluoranthene, and indeno[1,2,3-<i>c</i>,<i>d</i>]pyrene were studied and their adsorption behavior on carbon-based tweezer (CT) compound was estimated. Geometrical optimization based on DFT approach was considered, and molecular electrostatic potential (MEP) was mapped for further comparison between adsorption systems. Topological analysis such as reduced density gradient (RDG) in predicting the non-covalent interactions (NCI) at the adsorption surface was studied with a color map scale to characterize each interaction type. Absorbance evaluation through TD-DFT theory was involved to describe the optical behavior before and after adsorption. Molecular dynamic simulation was studied through 100 ps for some stability comparison between studied systems where temperature results full within 1250 to 1550 K for the four adsorption systems. The interaction on the CT surface was described by performing adsorption annealing module and the adsorption process mainly depended on the planar-like geometrical system for best surface interaction. The adsorption energy parameter for some estimated models can control the strength of interaction on the surface of CT compound. A benchmark study to compare the more suitable functional for adsorption systems description was performed using CAM-B3LYP/6-311++G functional and confirmed the more precise involved method with BBSE consideration.</p>","PeriodicalId":761,"journal":{"name":"Russian Journal of General Chemistry","volume":"94 10","pages":"2676 - 2688"},"PeriodicalIF":0.9000,"publicationDate":"2024-11-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Russian Journal of General Chemistry","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1134/S107036322410013X","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
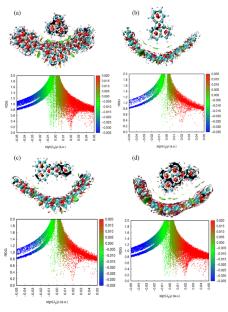
Polycyclic systems such as acenaphthylene, fluoranthene, benzo[k]fluoranthene, and indeno[1,2,3-c,d]pyrene were studied and their adsorption behavior on carbon-based tweezer (CT) compound was estimated. Geometrical optimization based on DFT approach was considered, and molecular electrostatic potential (MEP) was mapped for further comparison between adsorption systems. Topological analysis such as reduced density gradient (RDG) in predicting the non-covalent interactions (NCI) at the adsorption surface was studied with a color map scale to characterize each interaction type. Absorbance evaluation through TD-DFT theory was involved to describe the optical behavior before and after adsorption. Molecular dynamic simulation was studied through 100 ps for some stability comparison between studied systems where temperature results full within 1250 to 1550 K for the four adsorption systems. The interaction on the CT surface was described by performing adsorption annealing module and the adsorption process mainly depended on the planar-like geometrical system for best surface interaction. The adsorption energy parameter for some estimated models can control the strength of interaction on the surface of CT compound. A benchmark study to compare the more suitable functional for adsorption systems description was performed using CAM-B3LYP/6-311++G functional and confirmed the more precise involved method with BBSE consideration.
期刊介绍:
Russian Journal of General Chemistry is a journal that covers many problems that are of general interest to the whole community of chemists. The journal is the successor to Russia’s first chemical journal, Zhurnal Russkogo Khimicheskogo Obshchestva (Journal of the Russian Chemical Society ) founded in 1869 to cover all aspects of chemistry. Now the journal is focused on the interdisciplinary areas of chemistry (organometallics, organometalloids, organoinorganic complexes, mechanochemistry, nanochemistry, etc.), new achievements and long-term results in the field. The journal publishes reviews, current scientific papers, letters to the editor, and discussion papers.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


