Murugaiah A M Subbaiah, Thangeswaran Ramar, Maheswara Reddy, Sankara Sivaprasad Lvj, Shekhar Yeshwante, Srikanth Sridhar, Salil Desai, Manoj Chiney, Elizabeth A Dierks, Arvind Mathur, Ryan Moslin, David S Weinstein
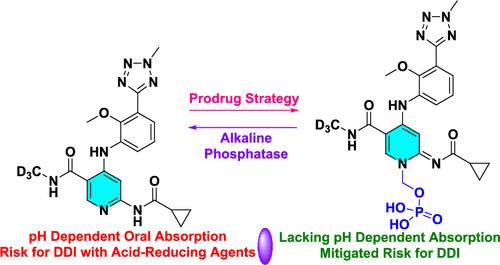
{"title":"Prodrug Strategy to Address Impaired Oral Absorption of a Weakly Basic TYK2 Inhibitor Caused by a Gastric Acid-Reducing Agent.","authors":"Murugaiah A M Subbaiah, Thangeswaran Ramar, Maheswara Reddy, Sankara Sivaprasad Lvj, Shekhar Yeshwante, Srikanth Sridhar, Salil Desai, Manoj Chiney, Elizabeth A Dierks, Arvind Mathur, Ryan Moslin, David S Weinstein","doi":"10.1021/acs.jmedchem.4c02219","DOIUrl":null,"url":null,"abstract":"<p><p>The pH-dependent solubility of the weakly basic TYK2 inhibitor <b>1</b> posed a risk to its advancement, given that drugs with such profiles have exhibited drug-drug interaction (DDI) with stomach acid-reducing agents in humans. In a rat model of pH dependence, preadministration of famotidine caused a 2.4-fold lower exposure of <b>1</b> when compared to control rats, implying that pH-dependent oral absorption can reduce the active drug's exposure and translate to subtherapeutic treatment. As part of risk mitigation, a prodrug strategy was explored by synthesizing solubility-enhancing prodrugs, resulting in the identification of lead prodrug <b>3c</b> with acceptable stability and solubility profiles. In rats, the prodrug eliminated the significant difference in AUC and <i>C</i><sub>max</sub> between pentagastrin and famotidine arms, thereby effectively mitigating the impaired drug absorption at the elevated pH relevant for absorption and DDI with famotidine. The prodrug also facilitated dose-proportional systemic exposure of <b>1</b> following dose escalation in rats and monkeys.</p>","PeriodicalId":46,"journal":{"name":"Journal of Medicinal Chemistry","volume":" ","pages":""},"PeriodicalIF":6.8000,"publicationDate":"2024-11-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Medicinal Chemistry","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1021/acs.jmedchem.4c02219","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
Abstract
The pH-dependent solubility of the weakly basic TYK2 inhibitor 1 posed a risk to its advancement, given that drugs with such profiles have exhibited drug-drug interaction (DDI) with stomach acid-reducing agents in humans. In a rat model of pH dependence, preadministration of famotidine caused a 2.4-fold lower exposure of 1 when compared to control rats, implying that pH-dependent oral absorption can reduce the active drug's exposure and translate to subtherapeutic treatment. As part of risk mitigation, a prodrug strategy was explored by synthesizing solubility-enhancing prodrugs, resulting in the identification of lead prodrug 3c with acceptable stability and solubility profiles. In rats, the prodrug eliminated the significant difference in AUC and Cmax between pentagastrin and famotidine arms, thereby effectively mitigating the impaired drug absorption at the elevated pH relevant for absorption and DDI with famotidine. The prodrug also facilitated dose-proportional systemic exposure of 1 following dose escalation in rats and monkeys.
期刊介绍:
The Journal of Medicinal Chemistry is a prestigious biweekly peer-reviewed publication that focuses on the multifaceted field of medicinal chemistry. Since its inception in 1959 as the Journal of Medicinal and Pharmaceutical Chemistry, it has evolved to become a cornerstone in the dissemination of research findings related to the design, synthesis, and development of therapeutic agents.
The Journal of Medicinal Chemistry is recognized for its significant impact in the scientific community, as evidenced by its 2022 impact factor of 7.3. This metric reflects the journal's influence and the importance of its content in shaping the future of drug discovery and development. The journal serves as a vital resource for chemists, pharmacologists, and other researchers interested in the molecular mechanisms of drug action and the optimization of therapeutic compounds.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


