Byung Do Lee, Deepak Subhash Gavali, Heejeong Kim, Seonghwan Kim, Min Young Cho, Kyunglim Pyo, Young-Kook Lee, Woon Bae Park and Kee-Sun Sohn
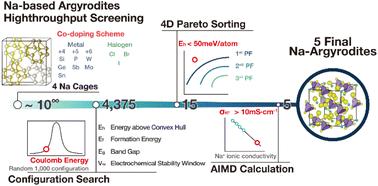
{"title":"Discovering virtual Na-based argyrodites as solid-state electrolytes using DFT, AIMD, and machine learning techniques†","authors":"Byung Do Lee, Deepak Subhash Gavali, Heejeong Kim, Seonghwan Kim, Min Young Cho, Kyunglim Pyo, Young-Kook Lee, Woon Bae Park and Kee-Sun Sohn","doi":"10.1039/D4TA06927G","DOIUrl":null,"url":null,"abstract":"<p >In surveying an extensive library of 4375 hypothetical Na-based argyrodites, we underscore the value of computational screening, noting that no Na-based argyrodite solid-state electrolyte has been successfully synthesized. We introduce a robust approach using density functional theory (DFT) calculations to identify thermodynamically and electrochemically stable candidates. By evaluating energy above the hull (<em>E</em><small><sub>h</sub></small>), formation energy (<em>E</em><small><sub>f</sub></small>), band gap (<em>E</em><small><sub>g</sub></small>), and electrochemical stability window (<em>V</em><small><sub>w</sub></small>), we narrow the set to 15 compounds <em>via</em> a 4-dimensional Pareto sorting. Competing materials for <em>E</em><small><sub>h</sub></small> and <em>V</em><small><sub>w</sub></small> calculations are sourced from the Materials Project, ICSD, and Google DeepMind. Connectivity-optimized graph networks validate the reliability of our calculations. <em>Ab initio</em> molecular dynamics (AIMD) calculations assess the room-temperature sodium ion conductivity (<em>σ</em><small><sub>RT</sub></small>) of the 15 selected entries, ultimately identifying the top 5 with promising <em>σ</em><small><sub>RT</sub></small>. This discovery of multi-compositional virtual argyrodites advances the challenge of synthesizing Na-based argyrodites.</p>","PeriodicalId":82,"journal":{"name":"Journal of Materials Chemistry A","volume":" 15","pages":" 10462-10474"},"PeriodicalIF":9.5000,"publicationDate":"2024-11-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Materials Chemistry A","FirstCategoryId":"88","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/ta/d4ta06927g","RegionNum":2,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
In surveying an extensive library of 4375 hypothetical Na-based argyrodites, we underscore the value of computational screening, noting that no Na-based argyrodite solid-state electrolyte has been successfully synthesized. We introduce a robust approach using density functional theory (DFT) calculations to identify thermodynamically and electrochemically stable candidates. By evaluating energy above the hull (Eh), formation energy (Ef), band gap (Eg), and electrochemical stability window (Vw), we narrow the set to 15 compounds via a 4-dimensional Pareto sorting. Competing materials for Eh and Vw calculations are sourced from the Materials Project, ICSD, and Google DeepMind. Connectivity-optimized graph networks validate the reliability of our calculations. Ab initio molecular dynamics (AIMD) calculations assess the room-temperature sodium ion conductivity (σRT) of the 15 selected entries, ultimately identifying the top 5 with promising σRT. This discovery of multi-compositional virtual argyrodites advances the challenge of synthesizing Na-based argyrodites.
期刊介绍:
The Journal of Materials Chemistry A, B & C covers a wide range of high-quality studies in the field of materials chemistry, with each section focusing on specific applications of the materials studied. Journal of Materials Chemistry A emphasizes applications in energy and sustainability, including topics such as artificial photosynthesis, batteries, and fuel cells. Journal of Materials Chemistry B focuses on applications in biology and medicine, while Journal of Materials Chemistry C covers applications in optical, magnetic, and electronic devices. Example topic areas within the scope of Journal of Materials Chemistry A include catalysis, green/sustainable materials, sensors, and water treatment, among others.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


