Shortcut to chemically accurate quantum computing via density-based basis-set correction
IF 5.9
2区 化学
Q1 CHEMISTRY, MULTIDISCIPLINARY
引用次数: 0
Abstract
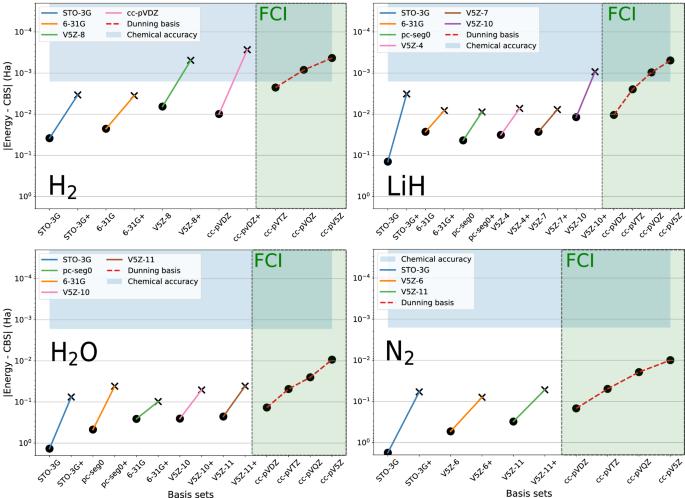
Using GPU-accelerated state-vector emulation, we propose to embed a quantum computing ansatz into density-functional theory via density-based basis-set corrections to obtain quantitative quantum-chemistry results on molecules that would otherwise require brute-force quantum calculations using hundreds of logical qubits. Indeed, accessing a quantitative description of chemical systems while minimizing quantum resources is an essential challenge given the limited qubit capabilities of current quantum processors. We provide a shortcut towards chemically accurate quantum computations by approaching the complete-basis-set limit through coupling the density-based basis-set corrections approach, applied to any given variational ansatz, to an on-the-fly crafting of basis sets specifically adapted to a given system and user-defined qubit budget. The resulting approach self-consistently accelerates the basis-set convergence, improving electronic densities, ground-state energies, and first-order properties (e.g. dipole moments), but can also serve as a classical, a posteriori, energy correction to quantum hardware calculations with expected applications in drug design and materials science. Quantum computing offers a promising approach to solving electronic-structure problems, but a quantitative description of chemical systems while minimizing computing resources is an essential challenge. Here, the authors provide a shortcut towards chemically accurate quantum computations by approaching the complete-basis-set limit through coupling the density-based basis-set corrections approach, applied to any given variational ansatz, to an on-the-fly crafting of basis sets specifically adapted to a given system and user-defined qubit budget.
通过基于密度的基集修正实现化学精确量子计算的捷径
利用 GPU 加速的状态矢量仿真,我们提议通过基于密度的基集修正,将量子计算解析嵌入密度函数理论,从而获得分子的定量量子化学结果,否则这些结果将需要使用数百个逻辑量子比特进行蛮力量子计算。事实上,鉴于当前量子处理器的量子比特能力有限,在获取化学系统定量描述的同时最大限度地减少量子资源是一项基本挑战。我们提供了一条实现化学精确量子计算的捷径,即通过将基于密度的基集修正方法(应用于任何给定的变分解析)与专门针对给定系统和用户定义的量子位预算的基集即时制作相结合,接近完整基集极限。由此产生的方法可以自洽地加速基集收敛,改善电子密度、基态能量和一阶属性(如偶极矩),还可以作为量子硬件计算的经典后验能量修正,有望应用于药物设计和材料科学领域。量子计算为解决电子结构问题提供了一种前景广阔的方法,但如何在定量描述化学系统的同时最大限度地减少计算资源是一项重大挑战。在此,作者提供了一条实现化学精确量子计算的捷径,即通过将基于密度的基集修正方法(适用于任何给定的变分解析式)与专门适应给定系统和用户定义的量子位预算的基集即时制作相结合,接近完全基集极限。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊
Communications Chemistry
Chemistry-General Chemistry
CiteScore
7.70
自引率
1.70%
发文量
146
审稿时长
13 weeks
期刊介绍:
Communications Chemistry is an open access journal from Nature Research publishing high-quality research, reviews and commentary in all areas of the chemical sciences. Research papers published by the journal represent significant advances bringing new chemical insight to a specialized area of research. We also aim to provide a community forum for issues of importance to all chemists, regardless of sub-discipline.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


