Effect of ordering of B’ site atom on the dynamical lattice properties of sustainable Sr2B′WO6 (B’= Co, Ni) double Perovskite
IF 3.8
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
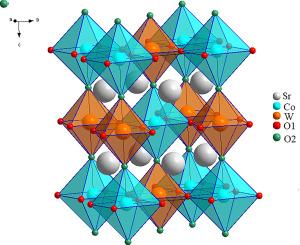
The wavenumbers of Raman and infrared spectra for environment friendly double perovskite Sr2B′WO6 (B’= Co, Ni) of I4/m phase (No.-87) has been analyzed using Wilson's GF matrix method. Theoretical assignments for the Raman and infrared wavenumbers for Sr2CoWO6 and Sr2NiWO6 compounds have been reported for the very first time. As far as we know, no theoretical assignments have been made for the infrared frequencies of Sr2CoWO6 and Sr2NiWO6 compounds in the I4/m phase. Both compounds are Lead-free oxide double perovskites which are emerging as a sustainable alternative to lead-based versions, offering similar electronic, optical, and magnetic properties without the associated environmental and health risks. A robust correlation with the B’ site has been identified across a range of frequencies, providing insights into the structural details of the examined compounds. The force constants related to the B’-site atom exhibit a consistent trend and reveal variations in values with changes in atomic orbitals. Moreover, frequencies influenced primarily by the B’ atoms display distinct characteristics with the variation of atomic number, emphasizing the considerable effect of the B’ atom's size on vibrational properties. The frequencies determined in this research align well with experimentally observed frequencies. Additionally, an exploration of potential energy distributions (PED) delves into the influence of interatomic forces on the computed Raman and infrared phonon modes for these materials.
B'位点原子有序化对可持续 Sr2B′WO6(B'= Co、Ni)双过氧化物动态晶格特性的影响
利用威尔逊 GF 矩阵法分析了 I4/m 相(No.-87)环保型双包晶 Sr2B′WO6(B'= Co、Ni)的拉曼光谱和红外光谱的波长。首次报告了 Sr2CoWO6 和 Sr2NiWO6 化合物的拉曼和红外波长的理论分配。据我们所知,还没有人对处于 I4/m 相的 Sr2CoWO6 和 Sr2NiWO6 化合物的红外频率进行过理论分配。这两种化合物都是无铅氧化物双包晶石,正在成为铅基化合物的可持续替代品,具有类似的电子、光学和磁学特性,但没有相关的环境和健康风险。在一定频率范围内确定了与 B'位点的稳健相关性,从而深入了解了所研究化合物的结构细节。与 B'位点原子相关的力常量呈现出一致的趋势,并揭示出其值随原子轨道变化而变化。此外,主要受 B'原子影响的频率随着原子序数的变化而显示出不同的特征,这突出了 B'原子的大小对振动特性的重要影响。这项研究确定的频率与实验观察到的频率非常吻合。此外,对势能分布(PED)的探索深入研究了原子间作用力对这些材料拉曼和红外声子模式计算结果的影响。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Chemical Physics Impact
Materials Science-Materials Science (miscellaneous)
CiteScore
2.60
自引率
0.00%
发文量
65
审稿时长
46 days
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


