Tuning the selectivity of Pd-based catalysts for CO oxidative esterification: Regulating Pd's electronic effect
IF 4.9
2区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
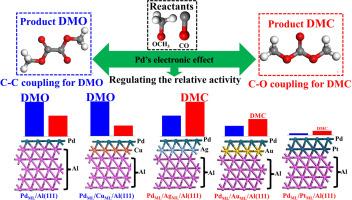
CO oxidative esterification is crucial in converting inorganic CO into organic oxygenated compounds within C1 chemistry and technology. However, understanding the structure-activity relationship underlying this reaction remains limited, primarily due to the entanglement of the Pd's geometric and electronic structures, which complicates selectivity control in practical applications. In this work, we design four Pd-based catalysts using the Al(111) motif to investigate their catalytic performance in CO oxidative esterification through density functional theory (DFT) calculations. Firstly, the larger lattice parameter of Al weakens horizontal Pd–Pd interactions while enhancing the vertical stability of Pd. Secondly, the electronic structure of Pd is locally regulated via charge transfer between subsurface and surface Pd, driven by their differences in work function. Our results indicate that the relative activity between C![]() C coupling for dimethyl oxalate (DMO) and C
C coupling for dimethyl oxalate (DMO) and C![]() O coupling for dimethyl carbonate (DMC) is primarily influenced by Pd's electronic effects, as revealed by energy barrier decomposition analysis. Thus, focusing on the regulation of Pd's electronic effects may offer a new strategy for controlling the selective oxidation esterification of CO.
O coupling for dimethyl carbonate (DMC) is primarily influenced by Pd's electronic effects, as revealed by energy barrier decomposition analysis. Thus, focusing on the regulation of Pd's electronic effects may offer a new strategy for controlling the selective oxidation esterification of CO.
调节 Pd 基催化剂在 CO 氧化酯化过程中的选择性:调节钯的电子效应
在 C1 化学和技术领域,一氧化碳氧化酯化是将无机一氧化碳转化为有机含氧化合物的关键。然而,人们对这一反应的结构-活性关系的了解仍然有限,这主要是由于钯的几何结构和电子结构之间存在纠缠,使得实际应用中的选择性控制变得复杂。在这项工作中,我们利用 Al(111) 主题设计了四种钯基催化剂,并通过密度泛函理论(DFT)计算研究了它们在 CO 氧化酯化反应中的催化性能。首先,Al 的较大晶格参数削弱了 Pd-Pd 的水平相互作用,同时增强了 Pd 的垂直稳定性。其次,钯的电子结构是通过表面下钯和表面钯之间的电荷转移进行局部调节的,这是由它们的功函数差异所驱动的。我们的研究结果表明,草酸二甲酯(DMO)的 CC 耦合和碳酸二甲酯(DMC)的 CO 耦合之间的相对活性主要受 Pd 电子效应的影响,能垒分解分析也揭示了这一点。因此,重点调节钯的电子效应可能会为控制 CO 的选择性氧化酯化提供一种新策略。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Molecular Catalysis
Chemical Engineering-Process Chemistry and Technology
CiteScore
6.90
自引率
10.90%
发文量
700
审稿时长
40 days
期刊介绍:
Molecular Catalysis publishes full papers that are original, rigorous, and scholarly contributions examining the molecular and atomic aspects of catalytic activation and reaction mechanisms. The fields covered are:
Heterogeneous catalysis including immobilized molecular catalysts
Homogeneous catalysis including organocatalysis, organometallic catalysis and biocatalysis
Photo- and electrochemistry
Theoretical aspects of catalysis analyzed by computational methods
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


