Jiangting Hu, Anna-Liisa Nieminen, Zhi Zhong, John J Lemasters
{"title":"Role of Mitochondrial Iron Uptake in Acetaminophen Hepatotoxicity.","authors":"Jiangting Hu, Anna-Liisa Nieminen, Zhi Zhong, John J Lemasters","doi":"10.3390/livers4030024","DOIUrl":null,"url":null,"abstract":"<p><p>Overdose of acetaminophen (APAP) produces fulminant hepatic necrosis. The underlying mechanism of APAP hepatotoxicity involves mitochondrial dysfunction, including mitochondrial oxidant stress and the onset of mitochondrial permeability transition (MPT). Reactive oxygen species (ROS) play an important role in APAP-induced hepatotoxicity, and iron is a critical catalyst for ROS formation. This review summarizes the role of mitochondrial ROS formation in APAP hepatotoxicity and further focuses on the role of iron. Normally, hepatocytes take up Fe<sup>3+</sup>-transferrin bound to transferrin receptors via endocytosis. Concentrated into lysosomes, the controlled release of iron is required for the mitochondrial biosynthesis of heme and non-heme iron-sulfur clusters. After APAP overdose, the toxic metabolite, NAPQI, damages lysosomes, causing excess iron release and the mitochondrial uptake of Fe<sup>2+</sup> by the mitochondrial calcium uniporter (MCU). NAPQI also inhibits mitochondrial respiration to promote ROS formation, including H<sub>2</sub>O<sub>2</sub>, with which Fe<sup>2+</sup> reacts to form highly reactive •OH through the Fenton reaction. •OH, in turn, causes lipid peroxidation, the formation of toxic aldehydes, induction of the MPT, and ultimately, cell death. Fe<sup>2+</sup> also facilitates protein nitration. Targeting pathways of mitochondrial iron movement and consequent iron-dependent mitochondrial ROS formation is a promising strategy to intervene against APAP hepatotoxicity in a clinical setting.</p>","PeriodicalId":74083,"journal":{"name":"Livers","volume":"4 3","pages":"333-351"},"PeriodicalIF":2.4000,"publicationDate":"2024-09-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11567147/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Livers","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3390/livers4030024","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/7/30 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
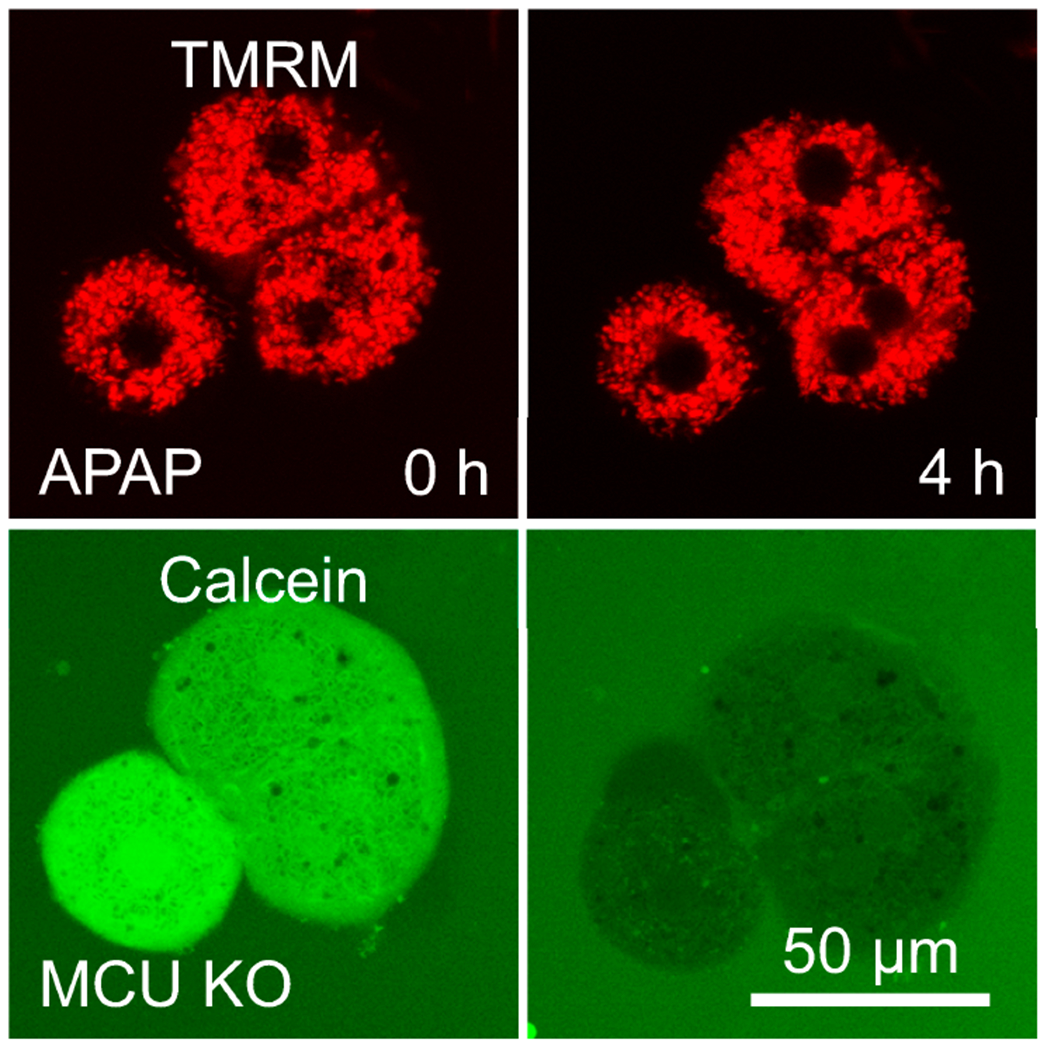
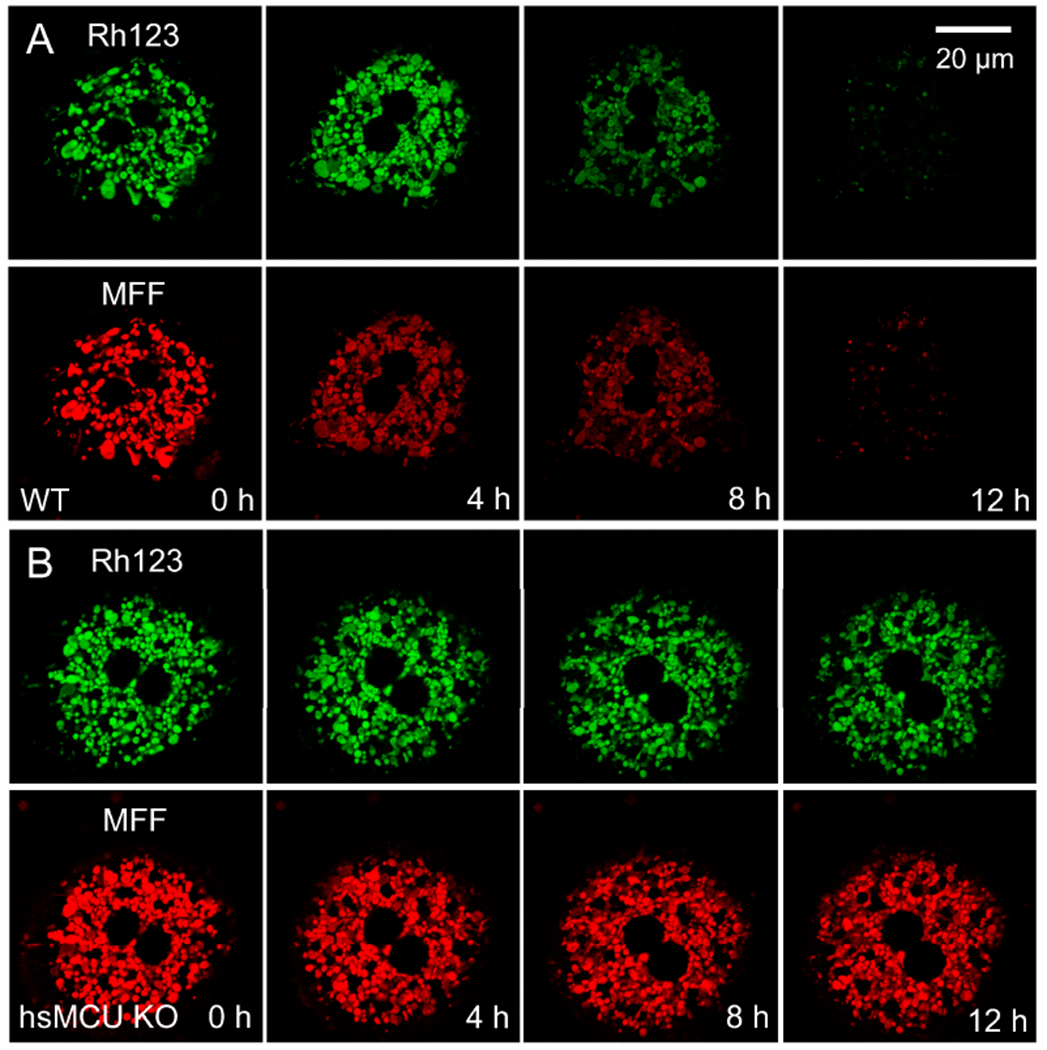
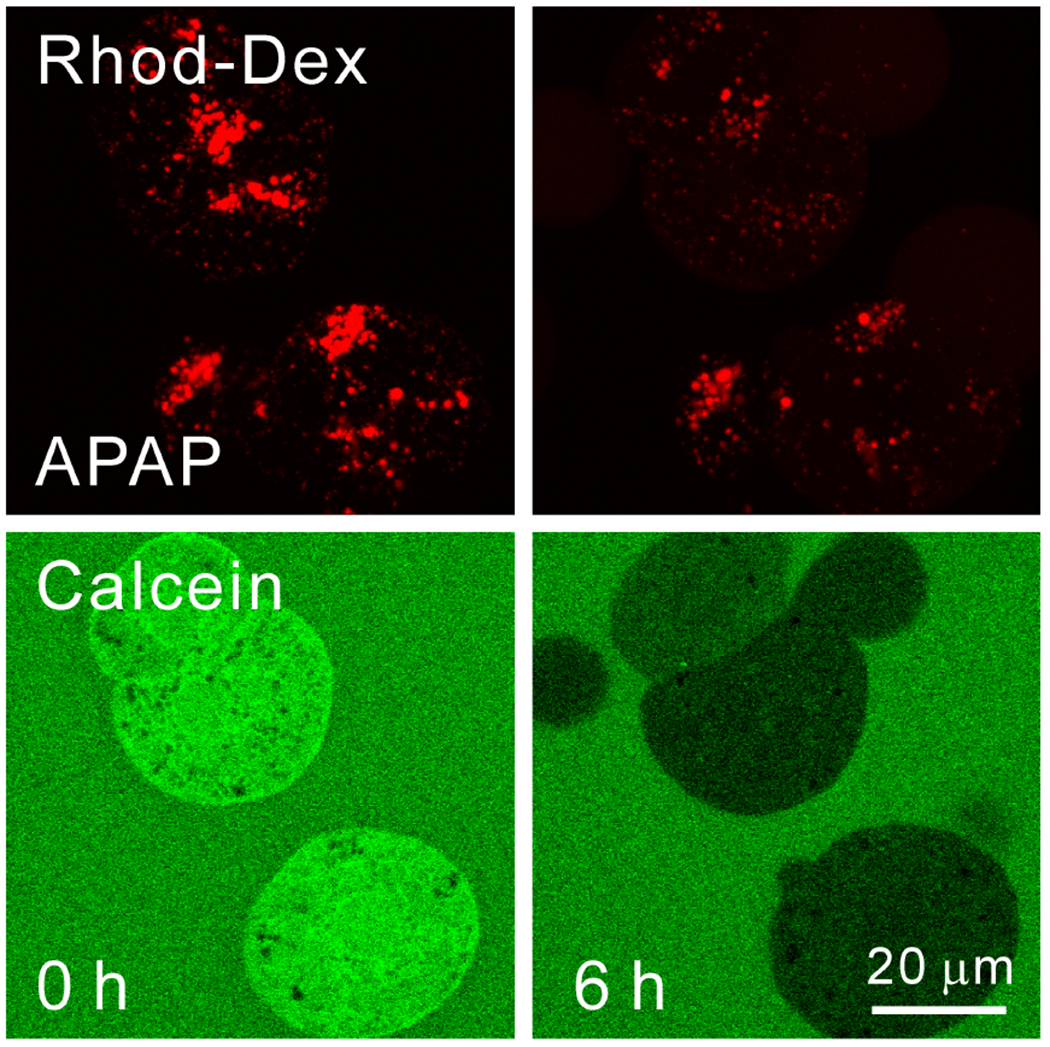
Overdose of acetaminophen (APAP) produces fulminant hepatic necrosis. The underlying mechanism of APAP hepatotoxicity involves mitochondrial dysfunction, including mitochondrial oxidant stress and the onset of mitochondrial permeability transition (MPT). Reactive oxygen species (ROS) play an important role in APAP-induced hepatotoxicity, and iron is a critical catalyst for ROS formation. This review summarizes the role of mitochondrial ROS formation in APAP hepatotoxicity and further focuses on the role of iron. Normally, hepatocytes take up Fe3+-transferrin bound to transferrin receptors via endocytosis. Concentrated into lysosomes, the controlled release of iron is required for the mitochondrial biosynthesis of heme and non-heme iron-sulfur clusters. After APAP overdose, the toxic metabolite, NAPQI, damages lysosomes, causing excess iron release and the mitochondrial uptake of Fe2+ by the mitochondrial calcium uniporter (MCU). NAPQI also inhibits mitochondrial respiration to promote ROS formation, including H2O2, with which Fe2+ reacts to form highly reactive •OH through the Fenton reaction. •OH, in turn, causes lipid peroxidation, the formation of toxic aldehydes, induction of the MPT, and ultimately, cell death. Fe2+ also facilitates protein nitration. Targeting pathways of mitochondrial iron movement and consequent iron-dependent mitochondrial ROS formation is a promising strategy to intervene against APAP hepatotoxicity in a clinical setting.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


