Felipe R. Dutra, João G. F. Romeu and David A. Dixon*,
{"title":"Prediction of Redox Potentials for Ac, Th, and Pa in Aqueous Solution","authors":"Felipe R. Dutra, João G. F. Romeu and David A. Dixon*, ","doi":"10.1021/acs.jpca.4c0569310.1021/acs.jpca.4c05693","DOIUrl":null,"url":null,"abstract":"<p >Density functional theory in conjunction with small core pseudopotentials and the associated basis sets was used to calculate potentials for multiple redox couples, covering a range of oxidation states for Ac (0 to III), Th (0 to IV), and Pa (0 to V) in aqueous solution. Solvation effects were incorporated using a supermolecule-continuum approach, with 30 water molecules representing two solvation shells, and the COSMO and SMD implicit solvation models. The calculated geometries for Ac(III), Th(IV), and Pa(V) were in reasonable agreement with the available experimental data. Using the COSMO model with the B3LYP functional, the calculated redox potentials were within ±0.2 V from experiment for most redox couples. Several pathways were explored for the Pa(V/IV) redox couple for different forms of Pa(V) and Pa(IV). Most Pa(V/IV) redox couples have very similar potentials, ranging from 0 to −0.4 V up to a pH of 1.4. At pH = 1.4, the potentials shift to values that are more negative than −0.7 V, reflecting the growing unfavorable nature of the redox process at higher pH levels. The calculated values for An(III/II) potentials were consistent with prior estimates and the available experimental data. The predicted redox potentials for An(II/I) were highly negative, as expected. For An(I/0) potentials, Th and Pa exhibited positive values, contrasting with the negative values calculated for Ac. The An<sup>+m</sup>/An(0) potentials agreed better with the experimental data when using the COSMO solvation model as compared to the SMD model.</p>","PeriodicalId":59,"journal":{"name":"The Journal of Physical Chemistry A","volume":"128 45","pages":"9730–9746 9730–9746"},"PeriodicalIF":2.7000,"publicationDate":"2024-10-31","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry A","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpca.4c05693","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
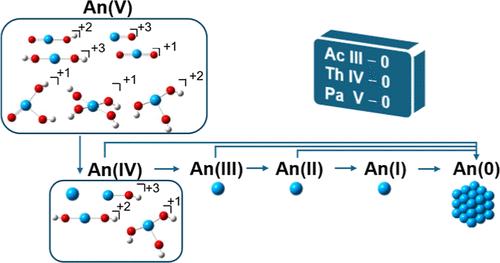
Density functional theory in conjunction with small core pseudopotentials and the associated basis sets was used to calculate potentials for multiple redox couples, covering a range of oxidation states for Ac (0 to III), Th (0 to IV), and Pa (0 to V) in aqueous solution. Solvation effects were incorporated using a supermolecule-continuum approach, with 30 water molecules representing two solvation shells, and the COSMO and SMD implicit solvation models. The calculated geometries for Ac(III), Th(IV), and Pa(V) were in reasonable agreement with the available experimental data. Using the COSMO model with the B3LYP functional, the calculated redox potentials were within ±0.2 V from experiment for most redox couples. Several pathways were explored for the Pa(V/IV) redox couple for different forms of Pa(V) and Pa(IV). Most Pa(V/IV) redox couples have very similar potentials, ranging from 0 to −0.4 V up to a pH of 1.4. At pH = 1.4, the potentials shift to values that are more negative than −0.7 V, reflecting the growing unfavorable nature of the redox process at higher pH levels. The calculated values for An(III/II) potentials were consistent with prior estimates and the available experimental data. The predicted redox potentials for An(II/I) were highly negative, as expected. For An(I/0) potentials, Th and Pa exhibited positive values, contrasting with the negative values calculated for Ac. The An+m/An(0) potentials agreed better with the experimental data when using the COSMO solvation model as compared to the SMD model.
期刊介绍:
The Journal of Physical Chemistry A is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


