Xiaoyang He, Li Lin, Xiangying Li, Minzhi Zhu, Qinghong Zhang, Shunji Xie, Bingbao Mei, Fanfei Sun, Zheng Jiang, Jun Cheng, Ye Wang
{"title":"Roles of copper(I) in water-promoted CO<sub>2</sub> electrolysis to multi-carbon compounds.","authors":"Xiaoyang He, Li Lin, Xiangying Li, Minzhi Zhu, Qinghong Zhang, Shunji Xie, Bingbao Mei, Fanfei Sun, Zheng Jiang, Jun Cheng, Ye Wang","doi":"10.1038/s41467-024-54282-2","DOIUrl":null,"url":null,"abstract":"<p><p>The membrane electrode assembly (MEA) is promising for practical applications of the electrocatalytic CO<sub>2</sub> reduction reaction (CO<sub>2</sub>RR) to multi-carbon (C<sub>2+</sub>) compounds. Water management is crucial in the MEA electrolyser without catholyte, but few studies have clarified whether the co-feeding water in cathode can enhance C<sub>2+</sub> formation. Here, we report our discovery of pivotal roles of a suitable nanocomposite electrocatalyst with abundant Cu<sub>2</sub>O-Cu<sup>0</sup> interfaces in accomplishing water-promoting effect on C<sub>2+</sub> formation, achieving a current density of 1.0 A cm<sup>-2</sup> and a 19% single-pass C<sub>2+</sub> yield at 80% C<sub>2+</sub> Faradaic efficiency in MEA. The operando characterizations confirm the co-existence of Cu<sup>+</sup> with Cu<sup>0</sup> during CO<sub>2</sub>RR at ampere-level current densities. Our studies reveal that Cu<sup>+</sup> works for water activation and aids C‒C coupling by enhancing formations of adsorbed CO and CHO species. This work offers a strategy to boost CO<sub>2</sub>RR to C<sub>2+</sub> compounds in industrial-relevant MEA by combining water management and electrocatalyst design.</p>","PeriodicalId":19066,"journal":{"name":"Nature Communications","volume":"15 1","pages":"9923"},"PeriodicalIF":14.7000,"publicationDate":"2024-11-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11568296/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nature Communications","FirstCategoryId":"103","ListUrlMain":"https://doi.org/10.1038/s41467-024-54282-2","RegionNum":1,"RegionCategory":"综合性期刊","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MULTIDISCIPLINARY SCIENCES","Score":null,"Total":0}
引用次数: 0
Abstract
The membrane electrode assembly (MEA) is promising for practical applications of the electrocatalytic CO2 reduction reaction (CO2RR) to multi-carbon (C2+) compounds. Water management is crucial in the MEA electrolyser without catholyte, but few studies have clarified whether the co-feeding water in cathode can enhance C2+ formation. Here, we report our discovery of pivotal roles of a suitable nanocomposite electrocatalyst with abundant Cu2O-Cu0 interfaces in accomplishing water-promoting effect on C2+ formation, achieving a current density of 1.0 A cm-2 and a 19% single-pass C2+ yield at 80% C2+ Faradaic efficiency in MEA. The operando characterizations confirm the co-existence of Cu+ with Cu0 during CO2RR at ampere-level current densities. Our studies reveal that Cu+ works for water activation and aids C‒C coupling by enhancing formations of adsorbed CO and CHO species. This work offers a strategy to boost CO2RR to C2+ compounds in industrial-relevant MEA by combining water management and electrocatalyst design.
期刊介绍:
Nature Communications, an open-access journal, publishes high-quality research spanning all areas of the natural sciences. Papers featured in the journal showcase significant advances relevant to specialists in each respective field. With a 2-year impact factor of 16.6 (2022) and a median time of 8 days from submission to the first editorial decision, Nature Communications is committed to rapid dissemination of research findings. As a multidisciplinary journal, it welcomes contributions from biological, health, physical, chemical, Earth, social, mathematical, applied, and engineering sciences, aiming to highlight important breakthroughs within each domain.
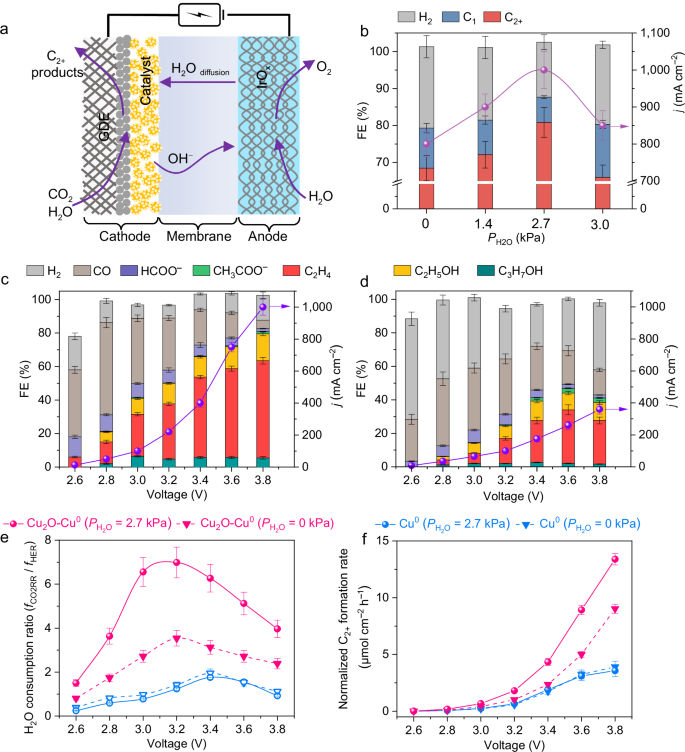

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


