{"title":"A Comparative Study of the Structural, Electronic, and Optical Properties of GaAs Phases Using Different Exchange-Correlation Functionals","authors":"Sadegh Mohammadpour Lima","doi":"10.1134/S1063783424601346","DOIUrl":null,"url":null,"abstract":"<p>We conducted first-principles calculations using density functional theory (DFT) to explore the structural, electronic, and optical characteristics of gallium arsenide (GaAs) in its wurtzite (WZ), zinc-blende (ZB), and rock-salt (RS) crystalline forms. These studies were carried out employing the full potential-linearized augmented plane wave (FP-LAPW) approach within the Wien2k computational framework. For the exchange-correlation (XC) potentials, we utilized various functionals including the local density approximation (LDA), generalized gradient approximation (PBE), Perdew–Burke–Ernzerhof for solids (PBEsol), modified Becke–Johnson (MBJ), and the strongly constrained and appropriately normed (SCAN) meta-generalized gradient approximation (meta-GGA). These functionals were applied to determine key physical properties such as equilibrium lattice constants, cell volume, bulk modulus, pressure derivatives, and energy band gaps. Additionally, we assessed optical features including dielectric and loss functions, refractive and extinction indices, and optical conductivity across the WZ, ZB, and RS phases. Our findings indicate that the structural and optical parameters derived from the SCAN meta-GGA functional show remarkable consistency with experimental observations, although it tends to underestimate the energy band gap. The most accurate predictions for the energy gap were achieved using the MBJ-PBEsol approach, aligning closely with experimental data.</p>","PeriodicalId":731,"journal":{"name":"Physics of the Solid State","volume":"66 11","pages":"543 - 555"},"PeriodicalIF":1.8000,"publicationDate":"2024-11-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physics of the Solid State","FirstCategoryId":"101","ListUrlMain":"https://link.springer.com/article/10.1134/S1063783424601346","RegionNum":4,"RegionCategory":"物理与天体物理","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"PHYSICS, CONDENSED MATTER","Score":null,"Total":0}
引用次数: 0
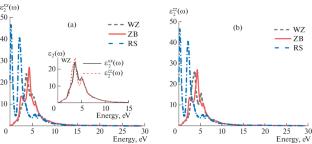
Abstract
We conducted first-principles calculations using density functional theory (DFT) to explore the structural, electronic, and optical characteristics of gallium arsenide (GaAs) in its wurtzite (WZ), zinc-blende (ZB), and rock-salt (RS) crystalline forms. These studies were carried out employing the full potential-linearized augmented plane wave (FP-LAPW) approach within the Wien2k computational framework. For the exchange-correlation (XC) potentials, we utilized various functionals including the local density approximation (LDA), generalized gradient approximation (PBE), Perdew–Burke–Ernzerhof for solids (PBEsol), modified Becke–Johnson (MBJ), and the strongly constrained and appropriately normed (SCAN) meta-generalized gradient approximation (meta-GGA). These functionals were applied to determine key physical properties such as equilibrium lattice constants, cell volume, bulk modulus, pressure derivatives, and energy band gaps. Additionally, we assessed optical features including dielectric and loss functions, refractive and extinction indices, and optical conductivity across the WZ, ZB, and RS phases. Our findings indicate that the structural and optical parameters derived from the SCAN meta-GGA functional show remarkable consistency with experimental observations, although it tends to underestimate the energy band gap. The most accurate predictions for the energy gap were achieved using the MBJ-PBEsol approach, aligning closely with experimental data.
期刊介绍:
Presents the latest results from Russia’s leading researchers in condensed matter physics at the Russian Academy of Sciences and other prestigious institutions. Covers all areas of solid state physics including solid state optics, solid state acoustics, electronic and vibrational spectra, phase transitions, ferroelectricity, magnetism, and superconductivity. Also presents review papers on the most important problems in solid state physics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


