Key Role of Positional Disorder and Soft Structural Framework for Lowering the Thermal Conductivity of Quaternary Ag1–xCu3+xTiSe4 (0 ≤ x ≤ 0.8) System to an Ultralow Limit
IF 4.4 2区 化学Q2 MATERIALS SCIENCE, MULTIDISCIPLINARY
{"title":"Key Role of Positional Disorder and Soft Structural Framework for Lowering the Thermal Conductivity of Quaternary Ag1–xCu3+xTiSe4 (0 ≤ x ≤ 0.8) System to an Ultralow Limit","authors":"Achintya Lakshan, Krishnendu Buxi, Paribesh Acharyya, Kishor Das, Biplab Koley, Kapildeb Dolui*, Christophe Candolfi, Carmelo Prestipino, Emmanuel Guilmeau, Ahin Roy and Partha Pratim Jana*, ","doi":"10.1021/acs.chemmater.4c0240410.1021/acs.chemmater.4c02404","DOIUrl":null,"url":null,"abstract":"<p >Low thermal conductive semiconductors have attracted huge attention for heat management and heat harvesting applications. Although the weak chemical bonding in the Cu/Ag-based chalcogenides is promising in suppressing heat transport, their quaternary analogs remain less explored. Here, we report on a comparative study of the crystal structure and thermal conductivity of various Ag-containing variants of Cu<sub>4</sub>TiSe<sub>4</sub>, i.e., Ag<sub>1–<i>x</i></sub>Cu<sub>3+<i>x</i></sub>TiSe<sub>4</sub> (<i>x</i> = 0–0.8) samples. Analysis of the crystal structure, phase transition, and temperature-dependent lattice thermal conductivity (κ<sub>L</sub>) of pristine AgCu<sub>3</sub>TiSe<sub>4</sub> and Ag<sub>1–<i>x</i></sub>Cu<sub>3+<i>x</i></sub>TiSe<sub>4</sub> have been carried out both experimentally and theoretically. The cubic crystal structure (space group <i>P</i>4̅3<i>m</i>) of these Ag variants is identical to that of Cu<sub>4</sub>TiSe<sub>4</sub> or Cu<sub>4</sub>TiTe<sub>4</sub>, where a positionally disordered Cu sublattice is either replaced by Ag (for AgCu<sub>3</sub>TiSe<sub>4</sub>) or by Ag/Cu substructure (for Ag<sub>1–<i>x</i></sub>Cu<sub>3+<i>x</i></sub>TiSe<sub>4</sub>). Upon cooling, the symmetry reduction to a rhombohedral (space group <i>R</i>3<i>m</i>) structure is attributed to the partial ordering of the positionally disordered Ag. The proposed structural models at different temperatures have been further analyzed using the Maximum Entropy Method (MEM). X-ray photoelectron spectroscopy measurement suggests that the parent compound forms a charge-precise (Ag<sup>+</sup>)(Cu<sup>+</sup>)<sub>3</sub>(Ti<sup>4+</sup>)(Se<sup>2–</sup>)<sub>4</sub> chemical formula. Interestingly, the lattice thermal conductivity of the Ag<sub>1–<i>x</i></sub>Cu<sub>3+<i>x</i></sub>TiSe<sub>4</sub> samples remains very low, with values varying in the range of ∼0.65–0.24 W m<sup>–1</sup> K<sup>–1</sup> between 293 and 623 K. Density Functional Theory (DFT) calculation shows the presence of antibonding states of Cu(3d)/Ag(4d)–Se(4p) below the Fermi level (<i>E</i><sub>F</sub>), providing softness to the lattice of AgCu<sub>3</sub>TiSe<sub>4</sub>. In addition, the positional disordered site plays a crucial role in further softening the framework and provides large lattice anharmonicity. The calculated phonon dispersions evidence the presence of several soft optical phonon modes at <i>ca.</i> 25 cm<sup>–1</sup>, originating from the atomic vibrations of Ag, Cu, and Se. Further confirmation of these phonon modes is obtained from the low-temperature heat capacity study. The low-lying optical phonon modes in AgCu<sub>3</sub>TiSe<sub>4</sub> are mainly caused by the presence of a soft lattice framework, positional disorder and associated rattling-like vibrations of Ag<sup>+</sup>/Cu<sup>+</sup> ions. Their strong interaction with the heat-carrying acoustic phonon modes is key ingredient that explains the very low κ<sub>L</sub>.</p>","PeriodicalId":7,"journal":{"name":"ACS Applied Polymer Materials","volume":null,"pages":null},"PeriodicalIF":4.4000,"publicationDate":"2024-10-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"ACS Applied Polymer Materials","FirstCategoryId":"88","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.chemmater.4c02404","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"MATERIALS SCIENCE, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
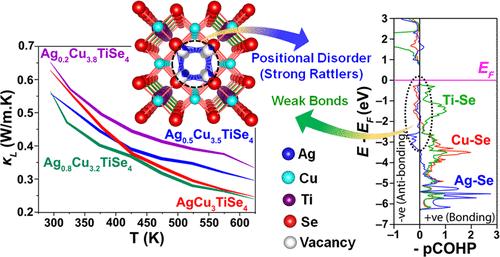
Low thermal conductive semiconductors have attracted huge attention for heat management and heat harvesting applications. Although the weak chemical bonding in the Cu/Ag-based chalcogenides is promising in suppressing heat transport, their quaternary analogs remain less explored. Here, we report on a comparative study of the crystal structure and thermal conductivity of various Ag-containing variants of Cu4TiSe4, i.e., Ag1–xCu3+xTiSe4 (x = 0–0.8) samples. Analysis of the crystal structure, phase transition, and temperature-dependent lattice thermal conductivity (κL) of pristine AgCu3TiSe4 and Ag1–xCu3+xTiSe4 have been carried out both experimentally and theoretically. The cubic crystal structure (space group P4̅3m) of these Ag variants is identical to that of Cu4TiSe4 or Cu4TiTe4, where a positionally disordered Cu sublattice is either replaced by Ag (for AgCu3TiSe4) or by Ag/Cu substructure (for Ag1–xCu3+xTiSe4). Upon cooling, the symmetry reduction to a rhombohedral (space group R3m) structure is attributed to the partial ordering of the positionally disordered Ag. The proposed structural models at different temperatures have been further analyzed using the Maximum Entropy Method (MEM). X-ray photoelectron spectroscopy measurement suggests that the parent compound forms a charge-precise (Ag+)(Cu+)3(Ti4+)(Se2–)4 chemical formula. Interestingly, the lattice thermal conductivity of the Ag1–xCu3+xTiSe4 samples remains very low, with values varying in the range of ∼0.65–0.24 W m–1 K–1 between 293 and 623 K. Density Functional Theory (DFT) calculation shows the presence of antibonding states of Cu(3d)/Ag(4d)–Se(4p) below the Fermi level (EF), providing softness to the lattice of AgCu3TiSe4. In addition, the positional disordered site plays a crucial role in further softening the framework and provides large lattice anharmonicity. The calculated phonon dispersions evidence the presence of several soft optical phonon modes at ca. 25 cm–1, originating from the atomic vibrations of Ag, Cu, and Se. Further confirmation of these phonon modes is obtained from the low-temperature heat capacity study. The low-lying optical phonon modes in AgCu3TiSe4 are mainly caused by the presence of a soft lattice framework, positional disorder and associated rattling-like vibrations of Ag+/Cu+ ions. Their strong interaction with the heat-carrying acoustic phonon modes is key ingredient that explains the very low κL.
期刊介绍:
ACS Applied Polymer Materials is an interdisciplinary journal publishing original research covering all aspects of engineering, chemistry, physics, and biology relevant to applications of polymers.
The journal is devoted to reports of new and original experimental and theoretical research of an applied nature that integrates fundamental knowledge in the areas of materials, engineering, physics, bioscience, polymer science and chemistry into important polymer applications. The journal is specifically interested in work that addresses relationships among structure, processing, morphology, chemistry, properties, and function as well as work that provide insights into mechanisms critical to the performance of the polymer for applications.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


