Orexin-A Attenuates the Inflammatory Response in Sepsis-Associated Encephalopathy by Modulating Oxidative Stress and Inhibiting the ERK/NF-κB Signaling Pathway in Microglia and Astrocytes
Abstract
Background
Oxidative stress-induced inflammation is a major pathogenic mechanism in sepsis-associated encephalopathy (SAE). We hypothesized that regulation of reactive oxygen species (ROS) by the neuropeptide orexin-A could prevent SAE-induced oxidative stress and inflammation. Therefore, the aim of this study was to investigate the effects of orexin-A on oxidative stress and inflammation in SAE in mice.
Methods
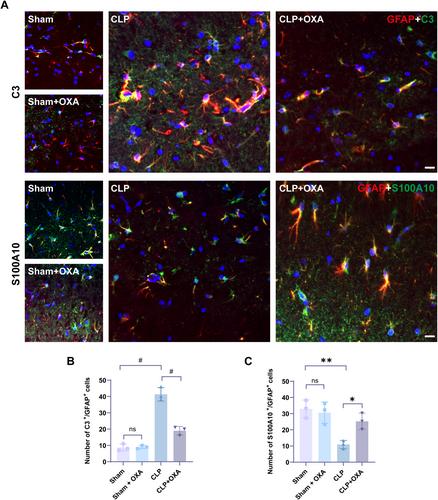
Adult male mice were treated with orexin-A (250 μg/kg, intranasal administration) to establish a cecal ligation perforation (CLP) model. We performed behavioral tests, observed neuronal damage in the hippocampal region, measured the levels of ROS, NOX2, and observed the structure of mitochondria by transmission electron microscopy. We then examined the inflammatory factors TNF-α and IL-1β, the activation of microglia and astrocytes, the expression of ERK/NF-κB, C3, and S100A10, and the presence of A1 type astrocytes and A2 type astrocytes.
Results
Orexin-A treatment improved cognitive performance in CLP-induced SAE mice, attenuated neuronal apoptosis in the hippocampal region, ameliorated ROS levels and the extent of mitochondrial damage, and reduced protein expression of NOX2 in hippocampal tissue. In addition, orexin-A treatment significantly reduced microglia and astrocyte activation, inhibited the levels of P-ERK and NF-κB, and reduced the release of IL-1β and TNF-α, which were significantly increased after CLP. Finally, Orexin-A treatment significantly decreased the number of C3/glial fibrillary acidic protein (GFAP)-positive cells and increased the number of S100A10/GFAP-positive cells.
Conclusion
Our data suggest that orexin-A reduces ROS expression by inhibiting CLP-induced NOX2 production, thereby attenuating mitochondrial damage and neuronal apoptosis. Its inhibition of microglial and A1-type astrocyte activation and inflammation was associated with the ERK/NF-κB pathway. These suggest that orexin-A may reduce cognitive impairment in SAE by reducing oxidative stress-induced inflammation.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


