Cross-comparison of gut metagenomic profiling strategies
IF 5.2
1区 生物学
Q1 BIOLOGY
引用次数: 0
Abstract
The rapid advancements in sequencing technologies and bioinformatics have enabled metagenomic research of complex microbial systems, but reliable results depend on consistent laboratory and bioinformatics approaches. Current efforts to identify best practices often focus on optimizing specific steps, making it challenging to understand the influence of each stage on microbial population analysis and compare data across studies. This study evaluated DNA extraction, library construction methodologies, sequencing platforms, and computational approaches using a dog stool sample, two synthetic microbial community mixtures, and various sequencing data sources. Our work, the most comprehensive evaluation of metagenomic methods to date. We developed a software tool, termed minitax, which provides consistent results across the range of platforms and methodologies. Our findings showed that the Zymo Research Quick-DNA HMW MagBead Kit, Illumina DNA Prep library preparation method, and the minitax bioinformatics tool were the most effective for high-quality microbial diversity analysis. However, the effectiveness of pipelines or method combinations is sample-specific, making it difficult to identify a universally optimal approach. Therefore, employing multiple approaches is crucial for obtaining reliable outcomes in microbial systems. Evaluation of metagenomic methods for gut microbiome profiling using dog stool and microbial standards, leading to the development of minitax tool. Optimal wetlab and analysis pipelines are recommended for reliable crossplatform microbiome studies.
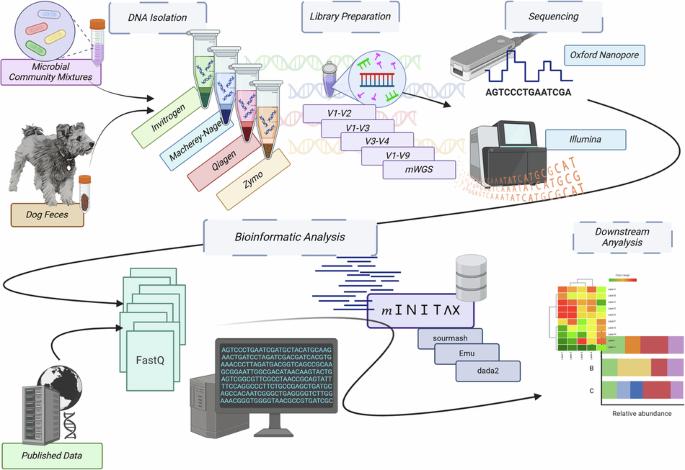
肠道元基因组剖析策略的交叉比较。
测序技术和生物信息学的快速发展使复杂微生物系统的元基因组研究成为可能,但可靠的结果取决于一致的实验室和生物信息学方法。目前,确定最佳实践的工作往往侧重于优化特定步骤,这使得了解每个阶段对微生物种群分析的影响以及比较不同研究的数据具有挑战性。本研究利用狗粪便样本、两种合成微生物群落混合物和各种测序数据源,对 DNA 提取、文库构建方法、测序平台和计算方法进行了评估。我们的工作是迄今为止对元基因组学方法最全面的评估。我们开发了一种称为 minitax 的软件工具,它能在各种平台和方法中提供一致的结果。我们的研究结果表明,Zymo Research Quick-DNA HMW MagBead Kit、Illumina DNA Prep 文库制备方法和 minitax 生物信息学工具对高质量微生物多样性分析最为有效。然而,管道或方法组合的有效性取决于具体样本,因此很难确定一种普遍适用的最佳方法。因此,要在微生物系统中获得可靠的结果,采用多种方法至关重要。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Communications Biology
Medicine-Medicine (miscellaneous)
CiteScore
8.60
自引率
1.70%
发文量
1233
审稿时长
13 weeks
期刊介绍:
Communications Biology is an open access journal from Nature Research publishing high-quality research, reviews and commentary in all areas of the biological sciences. Research papers published by the journal represent significant advances bringing new biological insight to a specialized area of research.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


